
- 首页 > 正文
肾例明鉴 | 两例IgG4相关性肾病合并单克隆丙种球蛋白病的诊疗解析及文献回顾
发表时间:2025-08-19 16:55:17
编者按:IgG4相关性疾病(IgG4-RD)是一种新分类的免疫介导的系统性疾病,目前我们对其认识尚浅。单克隆丙种球蛋白病是一组由克隆性浆细胞增殖引起的疾病,可导致血清或尿液中出现单克隆免疫球蛋白(M蛋白)。伴发单克隆丙种球蛋白病的IgG4相关性肾病较为罕见,其病因尚不明确。近期宁波市医疗中心李惠利医院肾内科董倩等发表文章,报告了2例罕见的IgG4相关性肾病合并单克隆丙种球蛋白病病例。本文予以介绍。
病例介绍
病例1
患者为中国男性,69岁,因肾功能异常于2023年10月17日入院检查。5个月前,患者出现夜尿增多,伴有泡沫尿、血尿、视力模糊、乏力及食欲减退,但无关节疼痛或下肢水肿。
实验室检查:血清肌酐为206.8 μmol/L,尿蛋白/肌酐比值(UPCR)为1.39 mg/mg。患者有6个多月的高血压病史和4年的贫血病史。体格检查未发现异常。
入院时的临床检查结果主要如下:嗜酸性粒细胞计数显著升高(300%);IgG和IgG4水平明显升高(分别为31.50 g/L和31.53 g/L);补体3(C3)和补体4(C4)显著降低(分别为38.90 mg/dl和1.67 mg/dl);游离轻链(FLC)κ为1021.98 mg/L,FLC λ为204.22 mg/L,κ/λ比值为5.00,存在单克隆IgG-λ蛋白;双侧腹股沟淋巴结肿大;肾脏超声显示双肾大小和形态正常,皮髓质分界清晰。患者进一步接受骨髓穿刺活检,结果显示有核细胞数量中度减少,淋巴细胞占18.5%,嗜酸性粒细胞占5.65%,未发现明显的原始和幼稚浆细胞。
进行常规超声引导下右肾活检以明确诊断。光镜下,6个肾小球呈微小病变,局灶性间质区域可见孤立的、富含胶原的严重纤维化(约40%面积)(图1A、B),可见“鸟眼征”和席纹状纤维化模式(图1C),并可见弥漫性炎症细胞浸润(主要为浆细胞,图1D)。免疫荧光显示IgG及其亚型、IgA、C3和C1q均为阴性,IgM(+/-)。肾间质中CD38和IgG4呈弥漫阳性;肾间质中IgG4强阳性,每高倍视野超过50个细胞,且IgG4/IgG比值>40%(图1E、F)。石蜡荧光显示:κ轻链少量弱阳性(平均10个细胞/HPF),λ轻链弥漫强阳性(平均100个细胞/HPF),两者分布差异显著(P<0.05)(图1G、H)。电镜显示:肾小球基底膜无明显病变,足突融合。诊断为IgG4相关性肾小管间质性肾炎(IgG4-TIN)合并具有肾脏意义的单克隆丙种球蛋白病(MGRS)。

图1
注:(A)轻度肾小球病变(PAS染色×400)。(B)富含胶原的孤立性纤维化(Masson染色×200)。(C)席纹状纤维化和“鸟眼征”改变,为IgG4-TIN的特异性形态学特征(PASM染色×400)。(D)大量肾间质炎症细胞(主要为浆细胞,伴有淋巴细胞、单核细胞和嗜酸性粒细胞)浸润(HE染色×200)。(E)IgG4阳性细胞数>50个/高倍视野(免疫组织化学×400)。(F)IgG阳性细胞,IgG4/IgG>40%(免疫组织化学×400)。(G)稀疏、弱阳性的κ轻链(石蜡荧光×200)。(H)弥漫、强阳性的λ轻链,提示单克隆淋巴浆细胞浸润(石蜡荧光×200)。
对患者初始给予甲泼尼龙40 mg/d静脉滴注,治疗3天后改为口服20 mg,随后逐渐减量。患者症状很快缓解。甲泼尼龙治疗开始后20周,血清肌酐逐渐降至94.10 μmol/L,白蛋白升至38.60 g/L,IgG和IgG4水平显著降低(分别为7.51 g/L和2.00 g/L),C3和C4水平显著升高(分别为110.20 mg/dl和28.00 mg/dl)。遗憾的是,由于医疗保险限制和患者经济状况,治疗后未能复查游离轻链水平(图2A)。未检测到单克隆蛋白,双侧腹股沟淋巴结恢复正常。
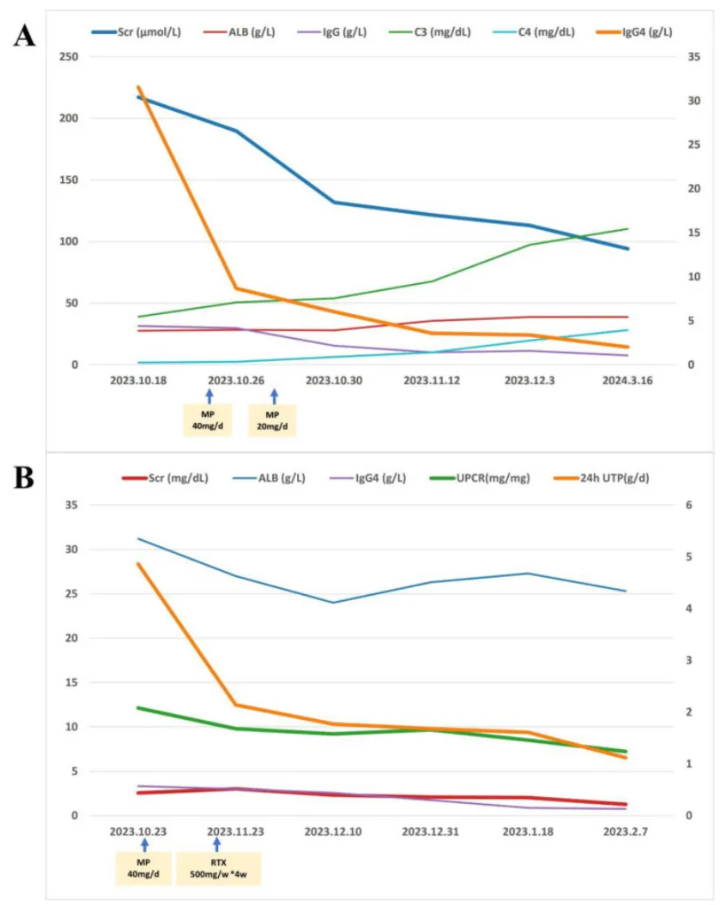
图2
注:(A)病例1接受甲泼尼龙静脉注射(40 mg/d,连续3天),随后口服甲泼尼龙(20 mg/d,逐渐减量)。随访20周时,观察到病情改善,包括血清肌酐(Scr)、IgG和IgG4水平降低,以及白蛋白(ALB)、补体C3和C4水平升高。(B)病例2接受甲泼尼龙静脉注射(40 mg/d,连续3天),随后口服甲泼尼龙(24 mg/d,逐渐减量),联合利妥昔单抗(每周500 mg,共4周)治疗。随访14周时,血清肌酐、24小时尿总蛋白(24hUTP)、尿蛋白/肌酐比值(UPCR)和IgG4水平降低,白蛋白水平稳定。
病例2
患者为中国男性,71岁,因高丙种球蛋白血症于2023年10月23日入院检查。1个月前,患者出现双下肢水肿,伴有泡沫尿,无皮疹、腹痛或腹泻。检查发现单克隆IgM-κ蛋白异常升高。6年前,患者曾患血栓性血小板减少性紫癜,经利妥昔单抗、糖皮质激素和静脉注射免疫球蛋白治疗成功。体格检查未发现异常。
入院时的临床检查结果主要汇总如下:血清肌酐225.10 μmol/L,24小时尿蛋白4858.60 mg/d,尿蛋白/肌酐比值2.08 mg/mg;IgG和IgG4水平显著升高(分别为44.70 g/L和3.32 g/L);游离轻链κ为287.59 mg/L,游离轻链λ为189.40 mg/L,κ/λ比值1.52,存在单克隆IgM-λ蛋白;磷脂酶A2受体(PLA2R)阴性;骨盆X线显示双侧髋关节退行性改变,可能双侧骶髂关节炎;肾脏超声显示双肾大小和形态正常,皮质回声增强,皮髓质分界不清;骨髓穿刺显示骨髓涂片易见嗜酸性粒细胞,成熟浆细胞占3.5%,无轻链限制性表达。
进行常规超声引导下右肾活检。光镜下,22个肾小球中13个呈全球性硬化,1个呈节段性硬化,1个有大型成纤维细胞性新月体。其余肾小球增大,基底膜弥漫增厚,伴有上皮下嗜品红蛋白沉积。PASM染色显示空泡变性(图3A、B)。肾间质可见多灶性炎症细胞浸润伴纤维化,无明显“鸟眼征”或席纹状纤维化(图3C、D)。免疫荧光显示:IgG(++)、IgG4(+)、IgA(+)、IgM(+)、C3(-)、C1q(+)、κ(+)、λ(+)沿毛细血管袢阳性沉积,其他IgG亚型、PLA2R和THSD7A均为阴性(图3E-G)。石蜡荧光显示:肾小球和间质中κ和λ阳性无显著差异。电镜下未观察到肾小球,肾小管基底膜未发现电子致密物沉积(图3H)。诊断为继发性膜性肾病(SMN)合并意义未明的单克隆丙种球蛋白病(MGUS),但不能排除IgG4相关性膜性肾病(IgG4-MN)。
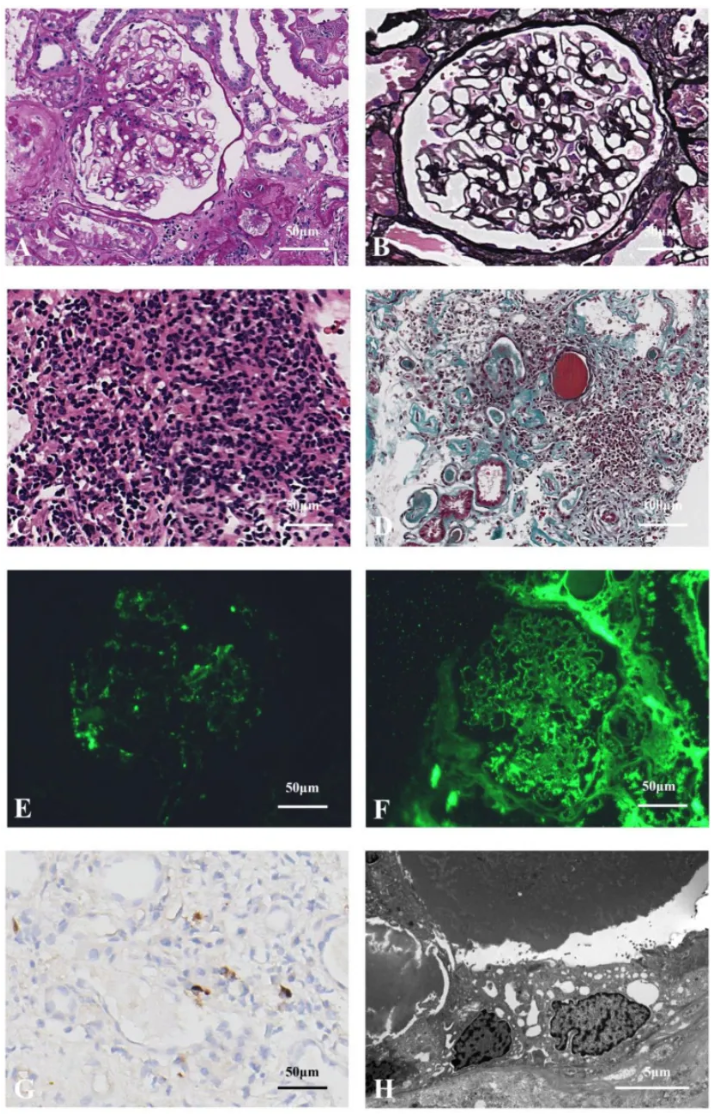
图3
注:(A)肾小球增大,基底膜增厚(PAS染色×400)。(B)肾小球基底膜增厚伴轻微空泡变性,为膜性肾病的特异性形态学特征(PASM染色×400)。(C)肾间质多灶性炎症细胞(主要为淋巴细胞)浸润(HE染色×400)。(D)肾间质炎症细胞浸润伴纤维化和肾小管萎缩,无席纹状特征(Masson染色×200)。(E)肾小球IgG4沿血管袢颗粒状阳性分布(石蜡荧光×400)。(F)肾小球IgG沿血管袢颗粒状阳性分布(石蜡荧光×400)。(G)肾间质中可见散在的IgG4阳性细胞(<10个/高倍视野),不符合IgG4-TIN的诊断标准(免疫组织化学×400)。(H)肾小管基底膜未发现电子致密物沉积(电镜×6000)。
对患者初始给予甲泼尼龙40 mg/d,治疗3天后改为口服24 mg,随后逐渐减量,联合利妥昔单抗(RTX,每周500 mg,共4周)治疗。治疗14周后,血清肌酐降至1.27 mg/dl,24小时尿蛋白降至1.12 g/d,IgG4水平恢复至正常范围,白蛋白未显著升高(图2B)。遗憾的是,由于医疗保险限制和患者经济状况,治疗后未能复查游离轻链水平。但M蛋白仍为IgM-λ型。
讨论
IgG4相关性疾病(IgG4-RD)是一种新定义的、伴有纤维化的免疫介导的慢性炎症性疾病。其主要病理表现包括以IgG4阳性浆细胞为主的淋巴浆细胞浸润、席纹状纤维化、闭塞性静脉炎和嗜酸性粒细胞浸润。关于IgG4-RD的全球发病率和患病率,已发表的数据有限。大多数患者为男性,年龄超过50岁,且在几周内对糖皮质激素治疗有反应。IgG4-RD的发病机制复杂,最近的研究发现CD4+细胞毒性T淋巴细胞(CTLs)发挥重要作用。此外,遗传风险和细菌感染也与IgG4-RD的发病机制有关。在IgG4相关性疾病(IgG4-RD)中,肾脏受累称为IgG4相关性肾病(IgG4-RKD)。最常见的表现是肾小管间质性肾炎(TIN),而膜性肾病(MN)是主要的肾小球病变,约占病例的7%。膜性肾病常与肾小管间质性肾炎并存,但也可单独出现。SaekiT等人(2020)制定了IgG4-RKD的综合诊断标准,包括肾功能、血清学、放射学和组织病理学参数,以规范临床诊断。IgG4-RKD的主要诊断标准包括:(1)肾脏表现(如急性或慢性肾功能不全、蛋白尿、低补体血症)和影像学表现(如双侧低回声肿块、弥漫性肾脏增大或肾盂肾盏壁增厚);(2)血清IgG4水平升高(通常>1.35 g/L);(3)组织病理学显示IgG4阳性浆细胞浸润(>10个/高倍视野)及特征性纤维化(席纹状纤维化或闭塞性静脉炎);(4)并发IgG4相关性疾病(如自身免疫性胰腺炎、泪腺炎)。病例1患者表现为肾功能不全、轻度蛋白尿、IgG4水平升高及淋巴结肿大,肾活检显示多灶性、片状肾间质纤维化,IgG4阳性细胞>10个/高倍视野,且IgG4/IgG>40%,根据诊断标准,可确诊为IgG4相关性肾小管间质性肾炎(IgG4-TIN)。病例2患者无IgG4系统性表现,肾组织中IgG4沉积较弱,不符合IgG4相关性肾病(IgG4-RKD)的诊断标准。
IgG4相关性疾病的临床表现高度多变,通常包括器官或组织肿胀、硬化,以及血清IgG4水平升高。诊断时需与炎症性、淋巴瘤性或恶性疾病相鉴别。回顾文献发现,有几例血清IgG4水平升高最终被诊断为淋巴瘤的病例报告。Ohta等人报告1例老年患者,血IgG4水平升高,腮腺和下颌下腺双侧肿胀,最终被诊断为边缘区淋巴瘤。经利妥昔单抗治疗后,患者下颌下腺肿胀改善,但血IgG4水平仍居高不下。Wu等人报告3例IgG4相关性眼病,最终进展为眼附属器边缘区B细胞淋巴瘤。同样,Nishida等人描述1例患者,最初接受糖皮质激素治疗,4年后被诊断为眼附属器边缘区B细胞淋巴瘤。HuiW等人最近报告1例IgG4相关性肾小管间质性肾炎合并间质炎症细胞λ轻链限制性表达的病例,最终被诊断为淋巴瘤。他们推测患者的弥漫性大B细胞淋巴瘤可能由先前存在但可能未被诊断的结外边缘区淋巴瘤转化而来。在病例1中,尽管在肾间质中观察到单克隆浆细胞浸润,但由于未进行淋巴结活检,尚不清楚这是否与淋巴结肿大相关。然而,随访结果显示,糖皮质激素治疗后IgG4水平显著下降,淋巴结大小恢复正常,肾功能改善,提示IgG4相关性肾病可能与M蛋白存在关联。长期预后,尤其是血液学变化,需要密切监测。病例2的肾活检结果显示肾小球增大,基底膜弥漫增厚,结合无IgG4相关性疾病的系统性表现、肾组织中IgG4沉积较弱及PLA2R染色阴性,倾向于诊断为继发性膜性肾病(SMN)。尽管如此,IgG4相关性膜性肾病(IgG4-MN)仍是可能的鉴别诊断。
单克隆丙种球蛋白病是浆细胞或B淋巴细胞的克隆性疾病,其特征是血清或尿液中可检测到M蛋白,范围从无症状疾病(如MGUS)到恶性肿瘤(如多发性骨髓瘤)。根据其临床意义和病因,单克隆丙种球蛋白病可分为以下类型:意义未明的单克隆丙种球蛋白病(MGUS)和有临床意义的单克隆丙种球蛋白病(MGCS)。MGUS通常无明显症状,可能在为其他疾病进行检查时偶然发现。其诊断标准包括:血清单克隆蛋白水平低于30 g/L;骨髓中浆细胞比例低于10%;无M蛋白相关的临床症状,如贫血、高钙血症、溶骨性病变或肾功能不全。中国人群中MGUS的患病率较高,尤其是在老年人中,50岁以上人群的患病率约为1.11%,70岁以上人群增至2.57%。西方国家的患病率略高,但差异不显著。MGUS的发病机制尚不清楚,但一般认为与遗传改变(如IgH易位、13号染色体缺失)、血管生成、细胞因子、骨髓瘤骨病和幽门螺杆菌有关。MGUS通常无需治疗,但由于存在转化为恶性浆细胞疾病的风险,需持续监测。如果患者符合MGUS的诊断标准且存在由M蛋白引起的肾脏损害,则应诊断为具有肾脏意义的单克隆丙种球蛋白病(MGRS)。本文中,两例患者均表现为单克隆丙种球蛋白病,但两例有所不同。病例1的肾组织中发现大量表达单克隆λ轻链的浆细胞,作者认为血液中的单克隆λ轻链已影响肾脏,因此诊断为MGRS。此外,糖皮质激素治疗后M蛋白消失,这可能归因于糖皮质激素的免疫抑制和抑制浆细胞分泌的作用。先前的一项研究也报告了1例具有肾脏意义的单克隆丙种球蛋白病亚型——伴M蛋白沉积的增生性肾小球肾炎(PGNMID),该病例对糖皮质激素治疗反应良好。相比之下,病例2的肾活检显示轻链表达无显著差异。经糖皮质激素和利妥昔单抗治疗后,蛋白尿改善,肾功能恢复,但血液中M蛋白持续存在。这表明血液中的M蛋白与肾脏损害无关,因此诊断为MGUS。MGUS和MGRS均可进展为恶性浆细胞疾病或淋巴增殖性疾病。因此,这两例患者未来需要密切的血液学随访。
总结
病例1诊断为IgG4相关性肾小管间质性肾炎合并具有肾脏意义的单克隆丙种球蛋白病,经糖皮质激素治疗后,肾脏和血液学指标均显著改善。病例2诊断为继发性膜性肾病合并意义未明的单克隆丙种球蛋白病,尽管经糖皮质激素和利妥昔单抗治疗后肾脏疾病有所改善,但M蛋白持续存在。通过这两例临床病例及对现有文献的回顾,本文阐明了IgG4相关性疾病的肾脏受累谱,强调了早期识别以防止不可逆纤维化的关键重要性。对于老年患者,加强血液学筛查并及时进行针对性干预,对于降低进展为多发性骨髓瘤等血液系统恶性肿瘤的风险至关重要。这些发现旨在帮助临床医生优化这种诊断不足疾病的诊断和治疗策略。
来源:Front Immunol. 2025 Jun 24:16:1539441. doi: 10.3389/fimmu.2025.1539441. eCollection 2025.
病例介绍
病例1
患者为中国男性,69岁,因肾功能异常于2023年10月17日入院检查。5个月前,患者出现夜尿增多,伴有泡沫尿、血尿、视力模糊、乏力及食欲减退,但无关节疼痛或下肢水肿。
实验室检查:血清肌酐为206.8 μmol/L,尿蛋白/肌酐比值(UPCR)为1.39 mg/mg。患者有6个多月的高血压病史和4年的贫血病史。体格检查未发现异常。
入院时的临床检查结果主要如下:嗜酸性粒细胞计数显著升高(300%);IgG和IgG4水平明显升高(分别为31.50 g/L和31.53 g/L);补体3(C3)和补体4(C4)显著降低(分别为38.90 mg/dl和1.67 mg/dl);游离轻链(FLC)κ为1021.98 mg/L,FLC λ为204.22 mg/L,κ/λ比值为5.00,存在单克隆IgG-λ蛋白;双侧腹股沟淋巴结肿大;肾脏超声显示双肾大小和形态正常,皮髓质分界清晰。患者进一步接受骨髓穿刺活检,结果显示有核细胞数量中度减少,淋巴细胞占18.5%,嗜酸性粒细胞占5.65%,未发现明显的原始和幼稚浆细胞。
进行常规超声引导下右肾活检以明确诊断。光镜下,6个肾小球呈微小病变,局灶性间质区域可见孤立的、富含胶原的严重纤维化(约40%面积)(图1A、B),可见“鸟眼征”和席纹状纤维化模式(图1C),并可见弥漫性炎症细胞浸润(主要为浆细胞,图1D)。免疫荧光显示IgG及其亚型、IgA、C3和C1q均为阴性,IgM(+/-)。肾间质中CD38和IgG4呈弥漫阳性;肾间质中IgG4强阳性,每高倍视野超过50个细胞,且IgG4/IgG比值>40%(图1E、F)。石蜡荧光显示:κ轻链少量弱阳性(平均10个细胞/HPF),λ轻链弥漫强阳性(平均100个细胞/HPF),两者分布差异显著(P<0.05)(图1G、H)。电镜显示:肾小球基底膜无明显病变,足突融合。诊断为IgG4相关性肾小管间质性肾炎(IgG4-TIN)合并具有肾脏意义的单克隆丙种球蛋白病(MGRS)。
图1
注:(A)轻度肾小球病变(PAS染色×400)。(B)富含胶原的孤立性纤维化(Masson染色×200)。(C)席纹状纤维化和“鸟眼征”改变,为IgG4-TIN的特异性形态学特征(PASM染色×400)。(D)大量肾间质炎症细胞(主要为浆细胞,伴有淋巴细胞、单核细胞和嗜酸性粒细胞)浸润(HE染色×200)。(E)IgG4阳性细胞数>50个/高倍视野(免疫组织化学×400)。(F)IgG阳性细胞,IgG4/IgG>40%(免疫组织化学×400)。(G)稀疏、弱阳性的κ轻链(石蜡荧光×200)。(H)弥漫、强阳性的λ轻链,提示单克隆淋巴浆细胞浸润(石蜡荧光×200)。
对患者初始给予甲泼尼龙40 mg/d静脉滴注,治疗3天后改为口服20 mg,随后逐渐减量。患者症状很快缓解。甲泼尼龙治疗开始后20周,血清肌酐逐渐降至94.10 μmol/L,白蛋白升至38.60 g/L,IgG和IgG4水平显著降低(分别为7.51 g/L和2.00 g/L),C3和C4水平显著升高(分别为110.20 mg/dl和28.00 mg/dl)。遗憾的是,由于医疗保险限制和患者经济状况,治疗后未能复查游离轻链水平(图2A)。未检测到单克隆蛋白,双侧腹股沟淋巴结恢复正常。
图2
注:(A)病例1接受甲泼尼龙静脉注射(40 mg/d,连续3天),随后口服甲泼尼龙(20 mg/d,逐渐减量)。随访20周时,观察到病情改善,包括血清肌酐(Scr)、IgG和IgG4水平降低,以及白蛋白(ALB)、补体C3和C4水平升高。(B)病例2接受甲泼尼龙静脉注射(40 mg/d,连续3天),随后口服甲泼尼龙(24 mg/d,逐渐减量),联合利妥昔单抗(每周500 mg,共4周)治疗。随访14周时,血清肌酐、24小时尿总蛋白(24hUTP)、尿蛋白/肌酐比值(UPCR)和IgG4水平降低,白蛋白水平稳定。
病例2
患者为中国男性,71岁,因高丙种球蛋白血症于2023年10月23日入院检查。1个月前,患者出现双下肢水肿,伴有泡沫尿,无皮疹、腹痛或腹泻。检查发现单克隆IgM-κ蛋白异常升高。6年前,患者曾患血栓性血小板减少性紫癜,经利妥昔单抗、糖皮质激素和静脉注射免疫球蛋白治疗成功。体格检查未发现异常。
入院时的临床检查结果主要汇总如下:血清肌酐225.10 μmol/L,24小时尿蛋白4858.60 mg/d,尿蛋白/肌酐比值2.08 mg/mg;IgG和IgG4水平显著升高(分别为44.70 g/L和3.32 g/L);游离轻链κ为287.59 mg/L,游离轻链λ为189.40 mg/L,κ/λ比值1.52,存在单克隆IgM-λ蛋白;磷脂酶A2受体(PLA2R)阴性;骨盆X线显示双侧髋关节退行性改变,可能双侧骶髂关节炎;肾脏超声显示双肾大小和形态正常,皮质回声增强,皮髓质分界不清;骨髓穿刺显示骨髓涂片易见嗜酸性粒细胞,成熟浆细胞占3.5%,无轻链限制性表达。
进行常规超声引导下右肾活检。光镜下,22个肾小球中13个呈全球性硬化,1个呈节段性硬化,1个有大型成纤维细胞性新月体。其余肾小球增大,基底膜弥漫增厚,伴有上皮下嗜品红蛋白沉积。PASM染色显示空泡变性(图3A、B)。肾间质可见多灶性炎症细胞浸润伴纤维化,无明显“鸟眼征”或席纹状纤维化(图3C、D)。免疫荧光显示:IgG(++)、IgG4(+)、IgA(+)、IgM(+)、C3(-)、C1q(+)、κ(+)、λ(+)沿毛细血管袢阳性沉积,其他IgG亚型、PLA2R和THSD7A均为阴性(图3E-G)。石蜡荧光显示:肾小球和间质中κ和λ阳性无显著差异。电镜下未观察到肾小球,肾小管基底膜未发现电子致密物沉积(图3H)。诊断为继发性膜性肾病(SMN)合并意义未明的单克隆丙种球蛋白病(MGUS),但不能排除IgG4相关性膜性肾病(IgG4-MN)。
图3
注:(A)肾小球增大,基底膜增厚(PAS染色×400)。(B)肾小球基底膜增厚伴轻微空泡变性,为膜性肾病的特异性形态学特征(PASM染色×400)。(C)肾间质多灶性炎症细胞(主要为淋巴细胞)浸润(HE染色×400)。(D)肾间质炎症细胞浸润伴纤维化和肾小管萎缩,无席纹状特征(Masson染色×200)。(E)肾小球IgG4沿血管袢颗粒状阳性分布(石蜡荧光×400)。(F)肾小球IgG沿血管袢颗粒状阳性分布(石蜡荧光×400)。(G)肾间质中可见散在的IgG4阳性细胞(<10个/高倍视野),不符合IgG4-TIN的诊断标准(免疫组织化学×400)。(H)肾小管基底膜未发现电子致密物沉积(电镜×6000)。
对患者初始给予甲泼尼龙40 mg/d,治疗3天后改为口服24 mg,随后逐渐减量,联合利妥昔单抗(RTX,每周500 mg,共4周)治疗。治疗14周后,血清肌酐降至1.27 mg/dl,24小时尿蛋白降至1.12 g/d,IgG4水平恢复至正常范围,白蛋白未显著升高(图2B)。遗憾的是,由于医疗保险限制和患者经济状况,治疗后未能复查游离轻链水平。但M蛋白仍为IgM-λ型。
讨论
IgG4相关性疾病(IgG4-RD)是一种新定义的、伴有纤维化的免疫介导的慢性炎症性疾病。其主要病理表现包括以IgG4阳性浆细胞为主的淋巴浆细胞浸润、席纹状纤维化、闭塞性静脉炎和嗜酸性粒细胞浸润。关于IgG4-RD的全球发病率和患病率,已发表的数据有限。大多数患者为男性,年龄超过50岁,且在几周内对糖皮质激素治疗有反应。IgG4-RD的发病机制复杂,最近的研究发现CD4+细胞毒性T淋巴细胞(CTLs)发挥重要作用。此外,遗传风险和细菌感染也与IgG4-RD的发病机制有关。在IgG4相关性疾病(IgG4-RD)中,肾脏受累称为IgG4相关性肾病(IgG4-RKD)。最常见的表现是肾小管间质性肾炎(TIN),而膜性肾病(MN)是主要的肾小球病变,约占病例的7%。膜性肾病常与肾小管间质性肾炎并存,但也可单独出现。SaekiT等人(2020)制定了IgG4-RKD的综合诊断标准,包括肾功能、血清学、放射学和组织病理学参数,以规范临床诊断。IgG4-RKD的主要诊断标准包括:(1)肾脏表现(如急性或慢性肾功能不全、蛋白尿、低补体血症)和影像学表现(如双侧低回声肿块、弥漫性肾脏增大或肾盂肾盏壁增厚);(2)血清IgG4水平升高(通常>1.35 g/L);(3)组织病理学显示IgG4阳性浆细胞浸润(>10个/高倍视野)及特征性纤维化(席纹状纤维化或闭塞性静脉炎);(4)并发IgG4相关性疾病(如自身免疫性胰腺炎、泪腺炎)。病例1患者表现为肾功能不全、轻度蛋白尿、IgG4水平升高及淋巴结肿大,肾活检显示多灶性、片状肾间质纤维化,IgG4阳性细胞>10个/高倍视野,且IgG4/IgG>40%,根据诊断标准,可确诊为IgG4相关性肾小管间质性肾炎(IgG4-TIN)。病例2患者无IgG4系统性表现,肾组织中IgG4沉积较弱,不符合IgG4相关性肾病(IgG4-RKD)的诊断标准。
IgG4相关性疾病的临床表现高度多变,通常包括器官或组织肿胀、硬化,以及血清IgG4水平升高。诊断时需与炎症性、淋巴瘤性或恶性疾病相鉴别。回顾文献发现,有几例血清IgG4水平升高最终被诊断为淋巴瘤的病例报告。Ohta等人报告1例老年患者,血IgG4水平升高,腮腺和下颌下腺双侧肿胀,最终被诊断为边缘区淋巴瘤。经利妥昔单抗治疗后,患者下颌下腺肿胀改善,但血IgG4水平仍居高不下。Wu等人报告3例IgG4相关性眼病,最终进展为眼附属器边缘区B细胞淋巴瘤。同样,Nishida等人描述1例患者,最初接受糖皮质激素治疗,4年后被诊断为眼附属器边缘区B细胞淋巴瘤。HuiW等人最近报告1例IgG4相关性肾小管间质性肾炎合并间质炎症细胞λ轻链限制性表达的病例,最终被诊断为淋巴瘤。他们推测患者的弥漫性大B细胞淋巴瘤可能由先前存在但可能未被诊断的结外边缘区淋巴瘤转化而来。在病例1中,尽管在肾间质中观察到单克隆浆细胞浸润,但由于未进行淋巴结活检,尚不清楚这是否与淋巴结肿大相关。然而,随访结果显示,糖皮质激素治疗后IgG4水平显著下降,淋巴结大小恢复正常,肾功能改善,提示IgG4相关性肾病可能与M蛋白存在关联。长期预后,尤其是血液学变化,需要密切监测。病例2的肾活检结果显示肾小球增大,基底膜弥漫增厚,结合无IgG4相关性疾病的系统性表现、肾组织中IgG4沉积较弱及PLA2R染色阴性,倾向于诊断为继发性膜性肾病(SMN)。尽管如此,IgG4相关性膜性肾病(IgG4-MN)仍是可能的鉴别诊断。
单克隆丙种球蛋白病是浆细胞或B淋巴细胞的克隆性疾病,其特征是血清或尿液中可检测到M蛋白,范围从无症状疾病(如MGUS)到恶性肿瘤(如多发性骨髓瘤)。根据其临床意义和病因,单克隆丙种球蛋白病可分为以下类型:意义未明的单克隆丙种球蛋白病(MGUS)和有临床意义的单克隆丙种球蛋白病(MGCS)。MGUS通常无明显症状,可能在为其他疾病进行检查时偶然发现。其诊断标准包括:血清单克隆蛋白水平低于30 g/L;骨髓中浆细胞比例低于10%;无M蛋白相关的临床症状,如贫血、高钙血症、溶骨性病变或肾功能不全。中国人群中MGUS的患病率较高,尤其是在老年人中,50岁以上人群的患病率约为1.11%,70岁以上人群增至2.57%。西方国家的患病率略高,但差异不显著。MGUS的发病机制尚不清楚,但一般认为与遗传改变(如IgH易位、13号染色体缺失)、血管生成、细胞因子、骨髓瘤骨病和幽门螺杆菌有关。MGUS通常无需治疗,但由于存在转化为恶性浆细胞疾病的风险,需持续监测。如果患者符合MGUS的诊断标准且存在由M蛋白引起的肾脏损害,则应诊断为具有肾脏意义的单克隆丙种球蛋白病(MGRS)。本文中,两例患者均表现为单克隆丙种球蛋白病,但两例有所不同。病例1的肾组织中发现大量表达单克隆λ轻链的浆细胞,作者认为血液中的单克隆λ轻链已影响肾脏,因此诊断为MGRS。此外,糖皮质激素治疗后M蛋白消失,这可能归因于糖皮质激素的免疫抑制和抑制浆细胞分泌的作用。先前的一项研究也报告了1例具有肾脏意义的单克隆丙种球蛋白病亚型——伴M蛋白沉积的增生性肾小球肾炎(PGNMID),该病例对糖皮质激素治疗反应良好。相比之下,病例2的肾活检显示轻链表达无显著差异。经糖皮质激素和利妥昔单抗治疗后,蛋白尿改善,肾功能恢复,但血液中M蛋白持续存在。这表明血液中的M蛋白与肾脏损害无关,因此诊断为MGUS。MGUS和MGRS均可进展为恶性浆细胞疾病或淋巴增殖性疾病。因此,这两例患者未来需要密切的血液学随访。
总结
病例1诊断为IgG4相关性肾小管间质性肾炎合并具有肾脏意义的单克隆丙种球蛋白病,经糖皮质激素治疗后,肾脏和血液学指标均显著改善。病例2诊断为继发性膜性肾病合并意义未明的单克隆丙种球蛋白病,尽管经糖皮质激素和利妥昔单抗治疗后肾脏疾病有所改善,但M蛋白持续存在。通过这两例临床病例及对现有文献的回顾,本文阐明了IgG4相关性疾病的肾脏受累谱,强调了早期识别以防止不可逆纤维化的关键重要性。对于老年患者,加强血液学筛查并及时进行针对性干预,对于降低进展为多发性骨髓瘤等血液系统恶性肿瘤的风险至关重要。这些发现旨在帮助临床医生优化这种诊断不足疾病的诊断和治疗策略。
来源:Front Immunol. 2025 Jun 24:16:1539441. doi: 10.3389/fimmu.2025.1539441. eCollection 2025.
- 推荐文章
肾例明鉴 | 老年男性ANCA相关性血管炎伴口干、眼干:只是非特异性抗体阳性?还是重叠综合征?——1例病例报告并文献综述
恩格列净+非奈利酮联用,高钾血症风险几何?CONFIDENCE试验二次分析结果揭晓
人人享有肾脏健康:关爱生命,守护地球——2026年世界肾脏日公益活动举行
14:00直播!2026年世界肾脏日公益活动即将开始——人人享有肾脏健康:关爱生命,守护地球
血液透析的环境挑战与可持续发展路径探索——2026世界肾脏日特别关注
特别策划 | 世界肾脏日:您真的了解肾脏健康吗?
肾域华章 | 儿童增殖性狼疮肾炎初始治疗添安全新选:多中心RCT证实吗替麦考酚酯非劣效于环磷酰胺选
2026世界肾脏日 | 绿色肾移植十策:让肾移植告别等待,共护双肾与地球
靶向清除“僵尸细胞”,达沙替尼联合槲皮素为糖尿病肾病治疗开辟新路径
慢性肾脏病与认知障碍:肾病越重,认知风险越高?——基于CRIC研究的新发现
膜性肾病精准诊疗的新证据:生物标志物和临床病理特征研究进展
eGFR百分位数:CKD早期识别的个性化新工具
PI解读:FSGS治疗新曙光——靶向药物迎来突破性进展
突破性进展:靶向补体因子B的RNA疗法为IgA肾病治疗带来新希望
利妥昔单抗血药浓度:膜性肾病早期治疗反应的预测标志物
肾例明鉴|43岁男子双下肢水肿、肌酐飙升,背后竟是两种肾病罕见叠加作祟!膜性肾病合并抗GBM病该如何破局?
Richard Lafayette教授:新型补体抑制剂时代下荚膜菌感染的防控指南
孙英贤教授牵头多中心研究:CRHCP事后分析揭秘CKM综合征不同分期强化降压价值
肾识百科|娃的肾病竟是遗传的?6种遗传性肾病家长必知!
护肾先护肠?1.2万人的迄今最大研究发现:肠道菌群及其代谢产物参与肾功能早期变化
肥胖与慢性肾脏病:不可忽视的“代谢多米诺”
《柳叶刀》:TRPC6抑制剂BI 764198治疗FSGS的Ⅱ期临床试验取得积极结果,为其足细胞靶向治疗提供首个有效证据
长期血透别忽视疼痛!平均8个部位受累,84%病程超1年、75%每日受折磨,肌肉骨骼和神经病理性痛为主要类型
近16万人数据:这样吃,大脑更年轻
美国麻省总医院最新综述:CKD-aP的诊疗进展与未来方向
超88%患者未获诊疗!Ⅲ期研究首证中国血透CKD-aP人群可从Difelikefalin治疗中获益
睡不好,血糖更难控?最新研究揭示:睡眠障碍正在悄悄扰乱你的“升糖”与“降糖”激素!
KDIGO肾小球疾病指南工作组主席Jürgen Floege教授:补体旁路途径是驱动CMKDs肾小球炎症的核心引擎
沉默的神偷!无声无息偷走小孩子肾脏功能的遗传性肾病——Dent病
国际组织在行动:CKM综合征、新型超级专科与“关爱肾脏”倡议
白蛋白尿——盐皮质激素受体拮抗剂带来肾脏获益的核心驱动因素
补体靶向时代:IgA肾病新疗法的疗效排名与精准化探索
从认知到实践:中国肾脏科医生IgA肾病诊疗现状与差距
肾例明鉴丨年轻痛风不是小事!19岁小伙关节痛3年,没控尿酸把肾“熬”成慢性肾病Ⅲ期
尿酸与非酒精性脂肪肝的“隐秘关联”:高血压人群需警惕的代谢信号
狼疮肾炎患者妊娠:她安然度过,她产后风暴骤起——来自两个临床病例研究的启示
罕见的先天性肾脏发育不良疾病——肾小球巨大而又稀少的“寡巨肾”,你听说过吗?
阿塞西普:IgA肾病治疗的新曙光——ORIGIN 3试验中期数据深度剖析
生殖史如何塑造年轻女性的心肾代谢风险?
指南共识丨《IgA肾病临床实践60问(2026版)》核心要点与临床实践解读(附原文)
司美格鲁肽获批治疗2型糖尿病合并慢性肾脏病1周年:糖尿病、肾脏与心脏疾病领域的变革
佳节健康“喝”出来——咖啡与茶,提神背后的健康双面性
春节期间重磅新闻速递:膜性肾病、IgA肾病、卵巢功能早衰领域迎来治疗新进展
“钾”在悄悄溜走,肾脏在喊“救命”!一种罕见的遗传性失盐性肾小管疾病
冬季假期真的会“悄悄让人发胖”吗?
肾友过年“四大护法”!作息·出行·娱乐·情绪,这份春节健康手册请收好
新春护肾,安心团圆:肾友及四高人群春节健康全攻略
学术纵横|多维靶向,精准护肾:小干扰RNA、ASI、nsMRA、GLP-1RA等新型药物均可为CKD患者保驾护航
尿糖阳性=糖尿病?那可不一定!这种罕见肾病会遗传尿糖,别当成普通糖尿病来治
聚焦CKM综合管理,恒格列净相关复方制剂赋能心-肾-代谢多重获益
年度盘点丨周晓玲教授:肥胖相关慢性肾脏病的诊治进展
肾例明鉴丨服药后肺肾接连“报警”!20岁女生的惊魂经历:元凶竟是治疗甲亢的丙硫氧嘧啶
谨慎“跟风养生”!Omega-3、生酮饮食、禁糖等营养宣传需辩证对待
肾识百科丨肾友想生娃?4类可怀情况 + 3类禁忌,提前看清不踩坑
警惕身体的“代谢紊乱风暴”!代谢综合征——引人关注的肾脏“杀手”