
- 首页 > 正文
肾例明鉴 | 夜尿2年竟藏致命血管炎!29岁男子的肾脏“嗜酸危机”,这个“沉默”的肾脏肉芽肿差点漏诊
发表时间:2026-04-25 14:43:55
编者按:变应性肉芽肿性血管炎(AGPA)是一种罕见的系统性小血管炎,典型表现为哮喘、嗜酸性粒细胞增多及多脏器损害三联征。临床确诊主要依赖临床表现与血清学检查(如ANCA),但该病发病率低且异质性强,极易漏诊。值得注意的是,由AGPA导致的慢性肾功能衰竭(CRF)并不多见,且当患者缺乏典型呼吸道症状或多系统受累时,诊断极具挑战性。此类不典型病例往往因无法满足常规诊断标准而被误诊为原发性肾小球疾病,导致错失免疫抑制治疗的最佳时机[1]。本文报道一例以恶性高血压及CRF为唯一表现的AGPA病例。患者无哮喘史,ANCA阴性,最终通过肾活检病理确诊。旨在通过分析该病例,强调在CRF病因筛查中,即使缺乏典型症状,也应高度警惕血管炎可能;同时指出肾活检对于明确病理本质、打破“多脏器受累”诊断刻板印象的关键价值。
1.病历摘要
患者,男性,29岁。因“夜尿增多2年,发现血压和血清肌酐明显升高1个月”入院。
2年前患者在无明显诱因下开始出现夜尿增多,2次/晚,但无其他症状,未作任何检查。1个月前因乏力、头昏、视物模糊,当地医院检查发现血压明显增高达160/110 mmHg,尿蛋白3+,尿RBC 0~1/HP,BUN 14.8 mmol/L,Scr 456.1 μmol/L。给予硝苯地平,西拉普利治疗,血压可以降至正常,但肾功能无好转,为进一步诊治收住入院。病程中无发热、哮喘、鼻炎、皮疹及关节痛等症状。发病后尿量1500~2000 ml/24h。
既往史、个人史与家族史无特殊。
体格检查:体温37.0℃,血压 左上肢140/90 mmHg,右上肢135/85 mmHg(服降压药后)。神志清楚,发育正常,营养中等,轻度贫血貌,全身无浮肿,皮肤无黄染及皮疹,感觉无异常。浅表淋巴结未触及,两肺呼吸音清,心律齐,各瓣膜听诊区均未闻及病理性杂音,腹壁柔软,肝脾肋下未触及,腹部未闻及血管杂音。四肢关节活动正常,动脉搏动无减弱,两侧对称。
辅助检查:尿液检查:蛋白定量3.54 g/24h,尿蛋白谱显示为混合性蛋白尿。尿RBC 55万/ml,多形型,嗜酸性粒细胞1100/ml,C3 14.0 mg/L。肾小管功能:溶菌酶>25 mg/L,NAG 25.1 U/(g·cr),禁水13h尿渗量345 mOsm/(kg·H2O)。肾小管酸化功能检查:pH 6.1,NAC 3 mmol/L,HCO3- 315 mmol/L,TA 8 mmol/L,NH4+ 10 mmol/L,NH4+/TA 1.25。血常规:Hb 95 g/L,WBC 5×109/L,N 64%,L 26%,嗜酸性粒细胞10%,嗜酸性粒细胞计数0.5×109/L,PLT 120×109/L。血生化:白蛋白31.9 g/L,球蛋白18.1 g/L,BUN 17.8 mmol/L,Scr 429.6 μmol/L,Ccr 13.43 ml/(min·1.73m²),钾4.6 mmol/L,钠140 mmol/L,氯111 mmol/L,TCO2 21 mmol/L,钙1.9 mmol/L,磷2.4 mmol/L,TC 5.4 mmol/L,TG 2.55 mmol/L,空腹血糖4.5 mmol/L,餐后2小时血糖6.5 mmol/L。免疫学:ANA、抗dsDNA抗体、抗Sm抗体、RF、ANCA(包括MPO-ANCA及PR3-ANCA)及AECA均为阴性。IgG 8.3 g/L、IgA 3.0 g/L、IgM 0.75 g/L、IgE 23.1 U/L,血清补体水平正常。冷球蛋白95 mg/L。大便检查:隐血试验阴性,未检出虫卵。
胸片:两肺纹理清晰,未见浸润性病灶。B超检查:肝内回声正常,血管纹路清晰,门静脉内径1.0 cm,胆囊及脾脏无异常;左肾96.1 mm×41.8 mm×43 mm,右肾91.2 mm×38.9 mm×41 mm,双侧肾上腺未见增大及占位。心电图:窦性心律。心动超声:左心室内径增大,无明显心包积液。血管彩色多普勒:肾内血流欠丰富,各血管峰值流速减低,各血管阻力指数PI、RI增高。骨髓常规:骨髓增生活跃,粒红各系细胞比例形态正常。眼底检查:动脉变细(A:V=1:3),反光增强,视盘旁片状出血,后极部见絮软性渗出,黄斑部硬性渗出。
肾活检病理:光镜:共10个肾小球,6个肾小球性废弃,其余肾小球细胞数明显减少,30~50个/球,基质增多,肾小球毛细血管袢塌陷、皱缩,大部分闭锁,偶见袢开放,数处假小管形成,包囊壁断裂。肾小管数量减少,残余肾小管萎缩、TBM增厚,部分扩张,内见巨大蛋白管型。间质弥漫嗜酸性粒细胞及单个核细胞浸润,嗜酸性粒细胞聚集成堆,形成肉芽肿样病变(图1)。小叶间动脉纤维素样坏死,血管壁增厚明显,部分管腔闭锁。肾间质小动脉血栓形成,内膜下见RBC碎片(图2)。肾间质见大量RBC。免疫病理:IgG++、IgA++、IgM++、C3++、C4和C1q+弥漫沉积于系膜区及血管袢。肾间质CD4 308/mm³,CD8 168/mm³,CD68 1016/mm³,PCNA 72/mm³,ICAM 细胞216/mm³。
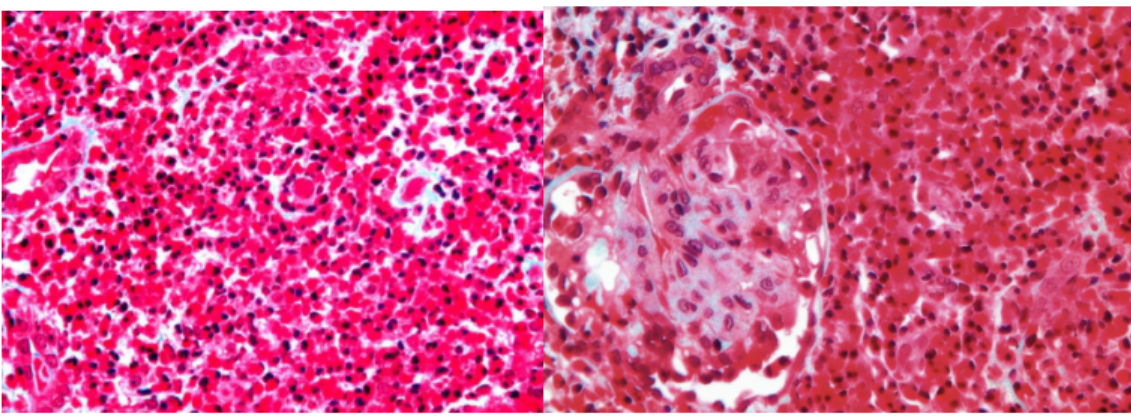
图1. 肾间质中大量嗜酸性粒细胞,呈“肉芽肿样”病变
入院后应用硝苯地平及苯那普利控制血压在130/80 mmHg。虽然患者肾组织已出现广泛纤维化,但仍有活动性血管炎、间质大量嗜酸性粒细胞及大量PCNA细胞浸润,表明病变仍处于活动,乃给予甲泼尼龙0.5 g静脉滴注,每日1次连续6天,此后改用口服泼尼松30 mg/d。甲泼尼龙冲击治疗后,患者尿量及血压无明显变化,但血及尿嗜酸性粒细胞迅速消失,BUN及Scr经过短期升高后缓慢下降,治疗2个月后Scr降至175 μmol/L,此外尿蛋白及尿RBC数量也明显减少。
最后诊断:变应性肉芽肿性血管炎;肾脏嗜酸性粒细胞性肉芽肿。
3.病例分析
本例患者主要表现为恶性高血压HBP及肾功能不全,尽管发现时间短,但B超显示双肾无肿大,结构不清,皮质回声增强,而且已有夜尿增多2年余,因此属于慢性肾功能衰竭(CRF)。患者有恶性HBP的表现,但在血压基本得到控制的情况下,仍持续存在大量蛋白尿,尿沉渣检查发现多形型RBC及大量嗜酸性粒细胞,表明肾脏损害不是单纯恶性HBP所致,而是存在肾小球病变,HBP为继发性。本例与某些原发性肾炎特别是临床有HBP的IgA肾病临床表现很相似,但有两点不符合。①尽管已经发生严重肾功能不全,但双肾体积并未明显缩小。②外周血及尿嗜酸性粒细胞增多,特别是尿嗜酸性粒细胞明显增多。
抓住嗜酸性粒细胞增多这一线索,CRF的病因需考虑。①过敏性间质性肾炎:单纯药物过敏性间质性肾炎,临床呈急性经过,往往伴有全身性症状如发热、皮疹、双肾增大,一般无严重HBP及大量蛋白尿,因而本病例不符合。但在原有肾脏病变的基础上合并过敏性间质性肾炎时,临床表现可类似本例,本例发病前无使用任何药物史,因此这种可能性不大,确诊依靠肾活检。②嗜酸性粒细胞白血病:白血病性嗜酸性粒细胞在肾组织大量浸润可以导致肾功能衰竭,但本病罕见,血嗜酸性粒细胞常达90%,骨髓嗜酸性粒细胞常>50%,肝(脾)大,肾脏也应增大,本例患者无这些特点。③胶原血管病:特别是系统性血管炎,临床往往有严重HBP及肾功能衰竭,肾脏大小与肾功能衰竭程度不平行。应首先考虑变应性肉芽肿性血管炎(allergic granulomatous with angiitis,AGPA,也曾称为Churg-Stuauss综合征),此外微型多动脉炎(MPA)及肉芽肿性血管炎(granulomatosis with polyangiitis,GPA,曾称为Wegener肉芽肿)也可以有严重肾功能衰竭及HBP,但一般不伴有嗜酸性粒细胞增多。MPA可以出现严重HBP、嗜酸性粒细胞增多,但不造成严重肾功能衰竭,除非有严重肾动脉狭窄[2,3]。这些疾病常有其他系统如消化道、神经系统、皮肤、关节及肺的受累,而本例患者无多脏器受损,临床证据不足,但不能排除不典型病例。单纯从临床表现无法确定诊断,血清学检查如ANCA也未能提供佐证,因此决定作肾活检。
肾活检组织学改变尽管显示大量肾小球硬化及广泛间质纤维化,但仍可见以下非常突出的病变。①肾间质小动脉坏死性血管炎。②血管外肉芽肿病变,嗜酸性粒细胞为主。③间质大量嗜酸性粒细胞浸润,但无肾小管炎。上述病理改变不符合过敏性间质性肾炎,而是提示存在一种伴有肉芽肿的血管炎。由于有肉芽肿形成,因此排除MPA,又有大量嗜酸性粒细胞浸润,GPA也可排除。结合外周血嗜酸性粒细胞增多,临床初步诊断为AGPA。与其他类型血管炎不同,激素治疗AGPA有较好疗效,本例患者经激素治疗后肾功能果然明显好转,进一步证明AGPA诊断正确。
AGPA是一种罕见的系统性小血管炎,临床发病率极低。多数患者肾脏损害轻微,严重肾功能衰竭者更为少见。由于临床表现不典型,该病存在较高的漏诊、误诊率,部分既往诊断为显微镜下MPA的患者实际可能为AGPA[4]。
本文报道的病例就呈现明显不典型特征:既无AGPA标志性的哮喘或过敏性鼻炎症状,血嗜酸性粒细胞仅轻度升高,也未出现多脏器损害,仅表现为肾脏受累。若未行肾活检获取病理证据,将无法按传统诊断标准确诊。
结合本病例的诊断,我们有以下临床体会:
1.CRF需明确病因:我国CRF以原发性肾小球肾炎最常见,但系统性血管炎等继发性病因所致肾功能衰竭可逆性较强,及时免疫抑制治疗可显著改善预后。临床需结合病史、实验室检查(如ANCA检测)及肾活检排查病因,避免延误病情。
2.AGPA诊断需结合临床与病理:典型AGPA分过敏性症状、嗜酸性粒细胞浸润、系统性血管炎三阶段,但部分病例表现不典型。其病理特征为坏死性小血管炎、血管外肉芽肿及嗜酸性粒细胞浸润,单次活检可能漏诊,需多部位、多次取材。ANCA对诊断有提示意义,但阴性不能排除AGPA。
3.动态认识多脏器损害特点:系统性血管炎病程漫长,不同阶段可能仅表现为单一脏器受累,经早期治疗后可不再进展。临床诊断不能拘泥于 “多脏器受累” 的刻板标准,需强调病理本质与动态随访。
总之,AGPA诊断无“金标准”,需综合临床、病理及动态观察,以减少漏诊误诊,改善患者预后。
参考文献:
[1] Narula N, Narula T, Derbes S, Espinoza LR. Churg-Strauss angiitis.Am J Med Sci. 2014;348(6):522-527.
[2] Abril A. Churg-strauss syndrome: an update. Curr Rheumatol Rep. 2011;13(6):489-495.
[3] Tobiáš D, Brázdilová K, Killinger Z, Payer J. Mikroskopická polyangiitída [Microscopic polyangiitis]. Vnitr Lek. 2020;66(4):249-252.
[4] Trivioli G, Marquez A, Martorana D, et al. Genetics of ANCA-associated vasculitis: role in pathogenesis, classification and management. Nat Rev Rheumatol. 2022;18(10):559-574.
1.病历摘要
患者,男性,29岁。因“夜尿增多2年,发现血压和血清肌酐明显升高1个月”入院。
2年前患者在无明显诱因下开始出现夜尿增多,2次/晚,但无其他症状,未作任何检查。1个月前因乏力、头昏、视物模糊,当地医院检查发现血压明显增高达160/110 mmHg,尿蛋白3+,尿RBC 0~1/HP,BUN 14.8 mmol/L,Scr 456.1 μmol/L。给予硝苯地平,西拉普利治疗,血压可以降至正常,但肾功能无好转,为进一步诊治收住入院。病程中无发热、哮喘、鼻炎、皮疹及关节痛等症状。发病后尿量1500~2000 ml/24h。
既往史、个人史与家族史无特殊。
体格检查:体温37.0℃,血压 左上肢140/90 mmHg,右上肢135/85 mmHg(服降压药后)。神志清楚,发育正常,营养中等,轻度贫血貌,全身无浮肿,皮肤无黄染及皮疹,感觉无异常。浅表淋巴结未触及,两肺呼吸音清,心律齐,各瓣膜听诊区均未闻及病理性杂音,腹壁柔软,肝脾肋下未触及,腹部未闻及血管杂音。四肢关节活动正常,动脉搏动无减弱,两侧对称。
辅助检查:尿液检查:蛋白定量3.54 g/24h,尿蛋白谱显示为混合性蛋白尿。尿RBC 55万/ml,多形型,嗜酸性粒细胞1100/ml,C3 14.0 mg/L。肾小管功能:溶菌酶>25 mg/L,NAG 25.1 U/(g·cr),禁水13h尿渗量345 mOsm/(kg·H2O)。肾小管酸化功能检查:pH 6.1,NAC 3 mmol/L,HCO3- 315 mmol/L,TA 8 mmol/L,NH4+ 10 mmol/L,NH4+/TA 1.25。血常规:Hb 95 g/L,WBC 5×109/L,N 64%,L 26%,嗜酸性粒细胞10%,嗜酸性粒细胞计数0.5×109/L,PLT 120×109/L。血生化:白蛋白31.9 g/L,球蛋白18.1 g/L,BUN 17.8 mmol/L,Scr 429.6 μmol/L,Ccr 13.43 ml/(min·1.73m²),钾4.6 mmol/L,钠140 mmol/L,氯111 mmol/L,TCO2 21 mmol/L,钙1.9 mmol/L,磷2.4 mmol/L,TC 5.4 mmol/L,TG 2.55 mmol/L,空腹血糖4.5 mmol/L,餐后2小时血糖6.5 mmol/L。免疫学:ANA、抗dsDNA抗体、抗Sm抗体、RF、ANCA(包括MPO-ANCA及PR3-ANCA)及AECA均为阴性。IgG 8.3 g/L、IgA 3.0 g/L、IgM 0.75 g/L、IgE 23.1 U/L,血清补体水平正常。冷球蛋白95 mg/L。大便检查:隐血试验阴性,未检出虫卵。
胸片:两肺纹理清晰,未见浸润性病灶。B超检查:肝内回声正常,血管纹路清晰,门静脉内径1.0 cm,胆囊及脾脏无异常;左肾96.1 mm×41.8 mm×43 mm,右肾91.2 mm×38.9 mm×41 mm,双侧肾上腺未见增大及占位。心电图:窦性心律。心动超声:左心室内径增大,无明显心包积液。血管彩色多普勒:肾内血流欠丰富,各血管峰值流速减低,各血管阻力指数PI、RI增高。骨髓常规:骨髓增生活跃,粒红各系细胞比例形态正常。眼底检查:动脉变细(A:V=1:3),反光增强,视盘旁片状出血,后极部见絮软性渗出,黄斑部硬性渗出。
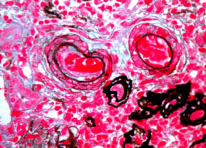
肾活检病理:光镜:共10个肾小球,6个肾小球性废弃,其余肾小球细胞数明显减少,30~50个/球,基质增多,肾小球毛细血管袢塌陷、皱缩,大部分闭锁,偶见袢开放,数处假小管形成,包囊壁断裂。肾小管数量减少,残余肾小管萎缩、TBM增厚,部分扩张,内见巨大蛋白管型。间质弥漫嗜酸性粒细胞及单个核细胞浸润,嗜酸性粒细胞聚集成堆,形成肉芽肿样病变(图1)。小叶间动脉纤维素样坏死,血管壁增厚明显,部分管腔闭锁。肾间质小动脉血栓形成,内膜下见RBC碎片(图2)。肾间质见大量RBC。免疫病理:IgG++、IgA++、IgM++、C3++、C4和C1q+弥漫沉积于系膜区及血管袢。肾间质CD4 308/mm³,CD8 168/mm³,CD68 1016/mm³,PCNA 72/mm³,ICAM 细胞216/mm³。
图1. 肾间质中大量嗜酸性粒细胞,呈“肉芽肿样”病变
图2. 肾间质小动脉血栓形成,内膜下见RBC碎片
入院后应用硝苯地平及苯那普利控制血压在130/80 mmHg。虽然患者肾组织已出现广泛纤维化,但仍有活动性血管炎、间质大量嗜酸性粒细胞及大量PCNA细胞浸润,表明病变仍处于活动,乃给予甲泼尼龙0.5 g静脉滴注,每日1次连续6天,此后改用口服泼尼松30 mg/d。甲泼尼龙冲击治疗后,患者尿量及血压无明显变化,但血及尿嗜酸性粒细胞迅速消失,BUN及Scr经过短期升高后缓慢下降,治疗2个月后Scr降至175 μmol/L,此外尿蛋白及尿RBC数量也明显减少。
最后诊断:变应性肉芽肿性血管炎;肾脏嗜酸性粒细胞性肉芽肿。
3.病例分析
本例患者主要表现为恶性高血压HBP及肾功能不全,尽管发现时间短,但B超显示双肾无肿大,结构不清,皮质回声增强,而且已有夜尿增多2年余,因此属于慢性肾功能衰竭(CRF)。患者有恶性HBP的表现,但在血压基本得到控制的情况下,仍持续存在大量蛋白尿,尿沉渣检查发现多形型RBC及大量嗜酸性粒细胞,表明肾脏损害不是单纯恶性HBP所致,而是存在肾小球病变,HBP为继发性。本例与某些原发性肾炎特别是临床有HBP的IgA肾病临床表现很相似,但有两点不符合。①尽管已经发生严重肾功能不全,但双肾体积并未明显缩小。②外周血及尿嗜酸性粒细胞增多,特别是尿嗜酸性粒细胞明显增多。
抓住嗜酸性粒细胞增多这一线索,CRF的病因需考虑。①过敏性间质性肾炎:单纯药物过敏性间质性肾炎,临床呈急性经过,往往伴有全身性症状如发热、皮疹、双肾增大,一般无严重HBP及大量蛋白尿,因而本病例不符合。但在原有肾脏病变的基础上合并过敏性间质性肾炎时,临床表现可类似本例,本例发病前无使用任何药物史,因此这种可能性不大,确诊依靠肾活检。②嗜酸性粒细胞白血病:白血病性嗜酸性粒细胞在肾组织大量浸润可以导致肾功能衰竭,但本病罕见,血嗜酸性粒细胞常达90%,骨髓嗜酸性粒细胞常>50%,肝(脾)大,肾脏也应增大,本例患者无这些特点。③胶原血管病:特别是系统性血管炎,临床往往有严重HBP及肾功能衰竭,肾脏大小与肾功能衰竭程度不平行。应首先考虑变应性肉芽肿性血管炎(allergic granulomatous with angiitis,AGPA,也曾称为Churg-Stuauss综合征),此外微型多动脉炎(MPA)及肉芽肿性血管炎(granulomatosis with polyangiitis,GPA,曾称为Wegener肉芽肿)也可以有严重肾功能衰竭及HBP,但一般不伴有嗜酸性粒细胞增多。MPA可以出现严重HBP、嗜酸性粒细胞增多,但不造成严重肾功能衰竭,除非有严重肾动脉狭窄[2,3]。这些疾病常有其他系统如消化道、神经系统、皮肤、关节及肺的受累,而本例患者无多脏器受损,临床证据不足,但不能排除不典型病例。单纯从临床表现无法确定诊断,血清学检查如ANCA也未能提供佐证,因此决定作肾活检。
肾活检组织学改变尽管显示大量肾小球硬化及广泛间质纤维化,但仍可见以下非常突出的病变。①肾间质小动脉坏死性血管炎。②血管外肉芽肿病变,嗜酸性粒细胞为主。③间质大量嗜酸性粒细胞浸润,但无肾小管炎。上述病理改变不符合过敏性间质性肾炎,而是提示存在一种伴有肉芽肿的血管炎。由于有肉芽肿形成,因此排除MPA,又有大量嗜酸性粒细胞浸润,GPA也可排除。结合外周血嗜酸性粒细胞增多,临床初步诊断为AGPA。与其他类型血管炎不同,激素治疗AGPA有较好疗效,本例患者经激素治疗后肾功能果然明显好转,进一步证明AGPA诊断正确。
AGPA是一种罕见的系统性小血管炎,临床发病率极低。多数患者肾脏损害轻微,严重肾功能衰竭者更为少见。由于临床表现不典型,该病存在较高的漏诊、误诊率,部分既往诊断为显微镜下MPA的患者实际可能为AGPA[4]。
本文报道的病例就呈现明显不典型特征:既无AGPA标志性的哮喘或过敏性鼻炎症状,血嗜酸性粒细胞仅轻度升高,也未出现多脏器损害,仅表现为肾脏受累。若未行肾活检获取病理证据,将无法按传统诊断标准确诊。
结合本病例的诊断,我们有以下临床体会:
1.CRF需明确病因:我国CRF以原发性肾小球肾炎最常见,但系统性血管炎等继发性病因所致肾功能衰竭可逆性较强,及时免疫抑制治疗可显著改善预后。临床需结合病史、实验室检查(如ANCA检测)及肾活检排查病因,避免延误病情。
2.AGPA诊断需结合临床与病理:典型AGPA分过敏性症状、嗜酸性粒细胞浸润、系统性血管炎三阶段,但部分病例表现不典型。其病理特征为坏死性小血管炎、血管外肉芽肿及嗜酸性粒细胞浸润,单次活检可能漏诊,需多部位、多次取材。ANCA对诊断有提示意义,但阴性不能排除AGPA。
3.动态认识多脏器损害特点:系统性血管炎病程漫长,不同阶段可能仅表现为单一脏器受累,经早期治疗后可不再进展。临床诊断不能拘泥于 “多脏器受累” 的刻板标准,需强调病理本质与动态随访。
总之,AGPA诊断无“金标准”,需综合临床、病理及动态观察,以减少漏诊误诊,改善患者预后。
参考文献:
[1] Narula N, Narula T, Derbes S, Espinoza LR. Churg-Strauss angiitis.Am J Med Sci. 2014;348(6):522-527.
[2] Abril A. Churg-strauss syndrome: an update. Curr Rheumatol Rep. 2011;13(6):489-495.
[3] Tobiáš D, Brázdilová K, Killinger Z, Payer J. Mikroskopická polyangiitída [Microscopic polyangiitis]. Vnitr Lek. 2020;66(4):249-252.
[4] Trivioli G, Marquez A, Martorana D, et al. Genetics of ANCA-associated vasculitis: role in pathogenesis, classification and management. Nat Rev Rheumatol. 2022;18(10):559-574.
- 推荐文章
ERA专家访谈丨顾乐怡教授:补体相关肾病治疗格局的范式跃迁,口服盐酸兰诺可泮治疗lgA肾病12周尿蛋白达标率高达40%
ERA中国之声 | 东南大学肾内科团队多项研究揭秘急性肾损伤全新作用机制
ERA中国之声丨UMOD H36Y突变如何通过代谢重编程加速肾小管衰老?
ERA专家访谈丨马坤岭教授:补体靶向治疗的“靶点前移”与“近端阻断”,口服盐酸兰诺可泮治疗IgA肾病12周尿蛋白达标率高达40%
ERA中国之声 | 重磅临床发现:抑酸药使用量与腹膜透析患者骨折风险呈梯度相关
ERA中国之声丨Dotinurad通过Nrf2介导的线粒体质量控制发挥直接肾小管保护作用,且该效应不依赖URAT1抑制
2026 ERA研究揭示从认知革新到实践破局的全球图景
江苏省中医院双项成果亮相ERA大会:靶向CTRP1-SGLT2与O-GlcNAc-FAO代谢记忆轴,破解糖尿病肾病发病新机制
ERA专家访谈丨路万虹教授:补体抑制剂助力IgA肾病早期、精准缓解,口服盐酸兰诺可泮治疗12周尿蛋白达标率高达40%
ERA中外专家研讨|聚焦IgA肾病前沿进展,解析临床对因治疗新看点
ERA 2026 | Crovalimab治疗成人及儿童aHUS:COMMUTE-a与COMMUTE-p Ⅲ期研究均达主要终点
ERA 2026 | 12周尿蛋白达标率高达40%,UPCR降幅51.8%!口服补体抑制剂盐酸兰诺可泮IgAN Ⅱ期高影响力研究数据全球首发
AI时代下聚焦批判性思维、技术创新与人文关怀!第63届ERA圆满闭幕,下一站鹿特丹
会议报道:从临床痛点出发,探寻IgA肾病长期管理最优解丨“肾英无界・中外IgA肾病学术前沿对话”成都站圆满收官
ERA 中国之声 | 华西医院30余项研究入选大会交流,4项研究上榜“青年作者优秀摘要”
全球首个APRIL靶向生物制剂斯贝利单抗在华获批,用于治疗IgA肾病
ERA 2026十佳摘要丨已用SGLT2i的2型糖尿病患者,联用GLP-1RA可再降27%肾病进展风险,全肾功能区间获益一致
ERA 2026前沿:三大新机制破局,CKD诊疗格局迎关键转折
ERA 2026重磅研究 | ALIGN试验2.5年eGFR结果:阿曲生坦治疗IgA肾病可显著、持续延缓肾功能下降
ERA 2026|9个月应答不佳仍具长期肾保护,NefIgArd亚组分析为IgA肾病对因治疗决策提供重磅证据【锁定今晚直播,洞悉2026 KDIGO更新要点】
ERA2026重磅研究丨FLOW试验最新数据:司美格鲁肽改善慢性肾脏病合并2型糖尿病患者生活质量
ERA 2026前沿 | 突破诊疗壁垒,迈向全程管理:构建以SGLT2i为核心的CKD早筛早诊早治新格局
ERA 2026十佳摘要丨中国台湾长庚医院单细胞图谱揭秘多囊肾发病源头,锁定全新靶向治疗靶点
ERA 2026丨突破9个月疗程,布地奈德肠溶胶囊长程治疗,为IgA肾病患者带来持久肾功能保护
ERA 2026 | 香港大学研究揭示:脂联素经线粒体重塑途径改善糖胖病相关肾损伤
杨淑敏教授:被忽视的20%——内分泌性高血压筛查专家共识解读
ERA 2026格拉斯哥正式启幕!以开放思维锚定肾病未来,《柳叶刀》重磅数据敲响CKD全球防控警钟
ERA 2026重磅丨IgA肾病尽早对因治疗:锚定蛋白尿≥0.5 g/d肾病进展风险人群,早用早达标、护肾更及时
ERA 2026前沿 | 突破“5.0时代”,迈向长期管理:真实世界研究驱动高钾血症诊疗变革
血液透析患者高血压管理的十个核心建议:基于ERA共识的临床实践指导
陈莉明教授:糖尿病慢性肾脏病的修正策略——从流行病学到多靶点治疗 | PUDF 2026
会议报道:聚焦临床痛点 共谋全程管理丨“肾英无界”中外IgA肾病学术前沿对话北京站成功举办
从AI到异种移植:ERA 2026最值得关注的五大未来技术
CIC 2026 | 任景怡教授:醛固酮介导心衰进展,nsMRA带来全新突破——从机制到临床的精准制导
ERA 2026三大临床试验专场:IgA肾病、CKD与罕见肾病治疗格局或将迎来新变化
何娅妮教授:糖尿病肾病治疗策略新进展——GDMT四大支柱筑牢心肾防线
Vadadustat(每周三次)对比不同基线剂量的甲氧基聚乙二醇促红素β治疗透析依赖型慢性肾病贫血患者的疗效与安全性分析
CCBPC 2026|涂晓文教授:血浆净化技术及其临床应用新进展
ERA 2026开幕在即!“Open your mind”背后,肾脏病学正在发生哪些改变?
ERA 2026|布地奈德肠溶胶囊23项全新研究闪耀登场,多维夯实对因、尽早、长期治疗临床证据体系
“生命之河”堵塞,肾脏告急!警惕“堵”出来的肾病——梗阻性肾病
中美双队列研究:不合并ASCVD的CKD患者,他汀治疗降低死亡风险26%~39%
会议报道:开放视野 探索前沿丨“肾英无界”中外IgA肾病学术前沿对话上海站圆满落幕
学术纵横丨常染色体显性多囊肾病(ADPKD)最新学术进展:风险评估、人群特征与创新药物探索
病例分享 | 血糖不高却频发酮症!这例糖尿病背后藏着双重隐秘病因,解密糖尿病胃轻瘫与肾糖阈减低的恶性循环
张宏教授:立足机制,解析B细胞靶向治疗IgA肾病的最新进展
肾移植术后膜性肾病研究:首个覆盖复发、未复发与新发类型的系统回顾与Meta分析核心结论与临床启示
压力与心血管疾病:成人期的隐形杀手
周晓霜教授:临床常规代谢指标与IgA肾病肾功能及病理损伤程度显著相关
非典型溶血尿毒综合征(aHUS)多学科诊疗专家共识要点,补体抑制剂为一线首选治疗
崔兆强教授:从“单纯降压”到“综合获益”——深度解析高血压治疗新趋势
以问破局 以共识远——《IgA肾病临床实践60问(2026版)》重磅发布,直击临床痛点,全面解锁IgA肾病对因治疗
开放思维,共探肾学前沿——第63届ERA大会即将启幕!26项中国研究入选最佳/优秀摘要
孙宁玲教授:破局“高容量”——从评估到干预,高血压管理如何走出困境?
CCBPC 2026|马坤岭教授:肠道菌群介导DKD肾损伤机制及转化应用新进展