
- 首页 > 正文
肾例明鉴丨从急肾衰到完全缓解:罕见多系统受累病例揭示 Castleman 病驱动抗GBM病的诊疗逻辑
发表时间:2026-03-13 16:40:31
Castleman病是一类以淋巴组织异常增殖为特征的罕见淋巴增殖性疾病,临床以单中心型多见,而多中心型浆细胞型Castleman病因常伴随全身炎症反应、多器官受累,诊疗难度更高,且易通过异常免疫激活诱发各类自身免疫并发症。抗肾小球基底膜病(抗GBM病)则是另一类罕见自身免疫性疾病,由抗GBM抗体介导,以急进性肾小球肾炎(RPGN)为典型表现,若未及时干预,多数患者短期内会进展为终末期肾病,预后极差。目前全球范围内,多中心型浆细胞型Castleman病合并抗GBM病的报道不足20例,二者的发病关联机制尚未完全阐明,临床诊疗中,此类患者多系统受累的复杂表现还易掩盖疾病本质,导致诊断延误与治疗方案偏差。本文将详细报道1例32岁男性患者Castleman病合并抗GBM病的诊疗过程,旨在为两种罕见疾病并存的临床诊疗提供参考。

一、临床资料
男性,32岁。主诉:发现血尿、蛋白尿1个月,血肌酐进行性升高2周。
现病史:1个月前无诱因发现尿色呈洗肉水样,伴双侧眼睑、下肢水肿,3周前于外院查血压140/90mmHg,尿常规:蛋白3+,潜血3+,尿蛋白定量7.2g/d,ALB 32g/L,SCr 118μmol/L,Hb 86g/L,ESR 60mm/h。2周后SCr升高至334μmol/L,尿量不少,900ml/d。收入院。
既往史:3年前患者搬入新房2个月后出现发热。午后明显,伴多汗,体温37~38℃,胸片、胸部CT及气管镜检查未发现异常。给予抗生素、抗结核治疗3~4个月,无效,半年后体温降至正常,此后未再发热。7个月前发现右侧颈部肿物,直径约3cm,无疼痛、发热,B超示双颈部、锁骨上及下颌多发淋巴结肿大,当地医院行淋巴结活检示“慢性淋巴结炎”,给予抗生素治疗,自觉肿物有所减小。患者为农民。吸烟10年,20支/天。
入院查体:体温37.0℃,脉搏80次/分,血压130/75mmHg,呼吸17次/分。双颈部、双下颌多发淋巴结肿大,最大者直径约4cm,边界清,无压痛。心肺查体未见异常。腹软,无压痛,未触及包块,肝肋下未触及,脾肋下两指,无压痛。双膝关节以下轻度凹陷性水肿。
辅助检查:血WBC 9.41×109/L,Hb 69g/L,PLT 84×109/L,ESR 107mm/h,CRP 78.1mg/dl。尿RBC 20~40/HP,形态正常,尿蛋白定量7.25g/d,ALB 27.2g/L,SCr 443μmol/L。肝功能正常。Anti-GBM 82%(正常<13%),ANCA(-)。IgG 21.1g/L,C3、C4正常。ANA(-)。Coombs试验(anti-IgG,anti-C3d)阳性。血小板相关抗体-IgG阴性。IL-6 401.8pg/ml(正常<100pg/ml),VEGF 209.5pg/ml(正常<29.1pg/ml)。B超:左肾14cm×6.0cm×4.8cm,实质厚1.9cm,右肾13.5cm×6.0cm×4.8cm,实质厚2.1cm,脂肪肝,胰腺周围偏低回声团——淋巴结?(直径1.2cm),脾大(厚5.9cm,长16.5cm)。双侧颈部多发肿大淋巴结,性质待定。胸片未见异常。
初步诊断:抗肾小球基底膜病;急进性肾小球肾炎Ⅰ型;自身免疫性溶血性贫血。淋巴结肿大原因待查
二、诊断思路和临床诊治经过
患者血肌酐短期内迅速升高,B超提示双肾体积增大、实质增厚,符合急性肾衰竭。根据患者出现血尿、大量蛋白尿、水肿及快速进展的肾功能损害,提示为肾小球疾病所致,符合急进性肾炎综合征。同时查血清抗GBM抗体阳性,因此诊断为抗GBM病,累及肾脏。
入院后行肾穿刺活检。免疫荧光:IgG(++),沿毛细血管壁线样沉积(图1)。光镜:22个肾小球,肾小球毛细血管袢严重破坏,其中2个节段性纤维素样坏死,11个细胞性、3个小细胞性新月体形成,其余系膜细胞和基质轻度弥漫增生,局灶节段性内皮细胞增生伴少量中性粒细胞浸润。肾小管上皮弥漫性刷状缘脱落,管腔内可见多数细胞管型。肾间质水肿,多灶状淋巴和单核细胞及多形核白细胞、嗜酸性粒细胞浸润。小动脉管壁增厚。符合新月体性肾炎,Ⅰ型。
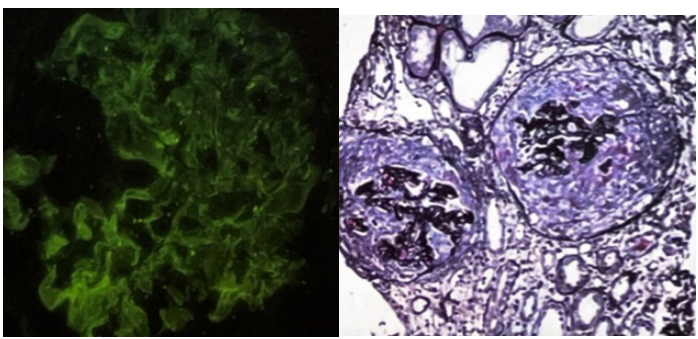
图1. 免疫荧光
患者同时存在全身多发的淋巴结肿大伴脾脏增大,原因不清。入院后再次行淋巴结活检病理:淋巴滤泡增生,生发中心毛细血管增生,可见无定型的嗜酸物质沉积,髓质多数浆细胞浸润,Mum1(+++),CD138(+++),符合巨大淋巴结增生症(Castleman病),浆细胞型(图2,图3)。因此诊断为多中心型浆细胞型Castleman病。在淋巴结活检组织中未检出人类疱疹病毒8。
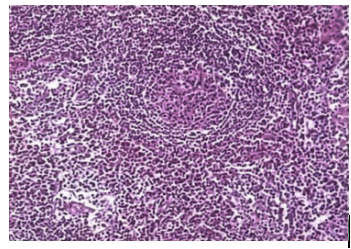
图2.淋巴索及淋巴窦内大浆细胞浸润
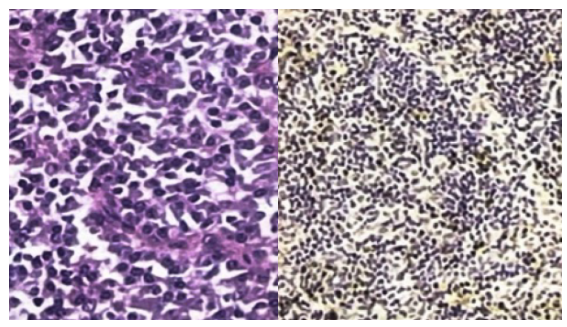
图3.大量浆细胞浸润,CD138阳性的浆细胞
患者入院后立即给予血浆置换治疗,每日1次,每次2000ml,共12次,血清抗GBM抗体转阴。同时给予甲泼尼龙500mg/d,共3天。2周后Coombs试验转阴,淋巴结和脾脏缩小,贫血及血小板减少均有所缓解。1个月后SCr、CRP和IL-6水平降至正常。其后患者每月接受1次COP方案化疗(环磷酰胺1.2g,长春新碱4mg和泼尼松100mg),6个月后获得完全缓解,期间监测抗GBM抗体始终为阴性。
最终诊断:抗肾小球基底膜病;急进性肾小球肾炎Ⅰ型;急性肾损伤;Castleman病多中心型浆细胞型。
二、诊断思路和临床诊治经过
这是一例Castleman病患者,同时出现由抗GBM抗体介导的急进性肾小球肾炎。给予强化血浆置换和免疫抑制治疗后,肾功能恢复,Castleman病也有所缓解。
Castleman病是一种少见的淋巴增殖性疾病。临床上分为单中心型和多中心型,病理上分为透明血管型、浆细胞型和混合细胞型。其中90%的病例为透明血管型,常无明显临床症状。浆细胞型可以出现系统症状,如发热、消瘦、皮疹、溶血性贫血和高丙种球蛋白血症[1]。
Castleman病的肾脏损害包括肾脏淀粉样变性、系膜增生性肾小球肾炎、血栓性微血管病、微小病变、间质性肾炎和膜性肾病,发病机制尚不清楚。新月体性肾炎的报道非常罕见,有报道抗MPO抗体相关,本例则与抗GBM抗体相关。
从流行病学角度看,Castleman病与抗GBM病均属于临床少见疾病,二者合并发生的报道更为罕见。Castleman病是一类以淋巴组织异常增生为特征的淋巴增殖性疾病,发病率约为 1/10万~2/10万,其中多中心型占比不足 30%,而浆细胞型在多中心型中占比仅10%~15%,常伴随全身炎症反应与多器官受累;抗GBM病则是由抗GBM抗体介导的自身免疫性疾病,年发病率约0.5/10万~1/10万,以急进性肾小球肾炎(Rapidly Progressive Glomerulonephritis,RPGN)为典型表现,若未及时治疗,90%以上患者会在 6 个月内进展为终末期肾病(End-Stage Renal Disease,ESRD)。截至目前,全球范围内关于Castleman病合并抗GBM病的报道不足 20 例,且多集中于单中心型Castleman病或混合细胞型Castleman病,本病例为多中心型浆细胞型Castleman病合并抗GBM病的少见实例,其临床价值在于进一步拓展了两种疾病关联的疾病谱,为探索自身免疫异常与淋巴增殖性疾病的相互作用机制提供了独特的临床样本[2,3]。
Castleman病也可以出现自身免疫系统异常,如获得性免疫性溶血性贫血、不典型系统性红斑狼疮,以及其他自身抗体,如抗核抗体、抗血小板抗体和抗心磷脂抗体等。这些多见于多中心型患者,单中心型少见。多克隆的B细胞活化导致出现高丙种球蛋白血症和自身抗体的产生。近期在Castleman病所致的一种严重的自身免疫性皮肤病变(副癌性天疱疮)中的研究发现,体外培养的Castleman肿瘤细胞能够分泌一种针对表皮蛋白的自身抗体[4]。本例患者的血清中也可检出抗GBM抗体,同时Coombs试验为阳性。随着Castleman病的缓解,上述抗体均转为阴性。进一步对该患者的淋巴结组织进行免疫荧光的染色,确实发现了能够分泌α3(Ⅳ)NC1抗体的浆细胞,从而证明了抗GBM抗体可由Castleman病肿瘤所活化的多克隆B细胞产生。
此外,本病例中IL-6水平的动态变化也为Castleman病的发病机制研究提供了重要参考。IL-6是Castleman病发病的核心细胞因子,正常生理状态下,IL-6主要由单核细胞、巨噬细胞分泌,参与炎症反应与免疫调节;而在浆细胞型Castleman病中,异常增殖的浆细胞与滤泡树突状细胞会大量分泌 IL-6,导致血清IL-6水平显著升高(正常参考值<100pg/ml),进而诱发全身炎症反应(如ESR、CRP升高)、浆细胞增殖与自身抗体产生。本病例患者入院时IL-6水平达401.8pg/ml,是正常上限的4倍,经血浆置换与化疗后,IL-6降至正常范围,同时伴随抗GBM抗体转阴、淋巴结缩小及肾功能恢复——这一动态变化不仅验证了IL-6在Castleman病病理生理过程中的核心作用,也提示IL-6可作为Castleman病病情活动度的重要监测指标,为后续Castleman病患者的病情评估与治疗反应判断提供了可靠的实验室依据[5]。
原发性抗GBM病的复发非常罕见,不需要给予持续性的免疫抑制治疗。本例患者由于药物副作用,经过6个月的规律化疗后停止治疗。之后其血清的IL-6水平再次升高,抗GBM抗体再次出现,这也提示抗体的出现是由Castleman病所致。因此对于Castleman病所致的抗GBM病应给予持续而规律的化学治疗。
综上,本病例通过规范的诊疗流程,实现了Castleman病与抗GBM病的同步缓解,不仅为临床医生提供了复杂病例的诊疗思路,也为两种罕见疾病的关联机制研究奠定了基础。未来需进一步加强多学科协作(肾内科、血液科、病理科),推动罕见病诊疗水平的提升,为患者提供更优质的医疗服务。
参考文献:
[1] Castleman Disease Collaborative Network (CDCN). International, evidence - based consensus diagnostic criteria for idiopathic multicentric Castleman disease. Blood. 2017;129 (13):1744 - 1754.
[2] Stone JH, et al. KDIGO Clinical Practice Guideline for Glomerulonephritis. Kidney Int Suppl. 2021;11 (1):S1 - S276.
[3] Jiang X, et al. Epidemiology, clinical features, risk factors, and outcomes of anti - glomerular basement membrane disease: A systematic review and meta - analysis. Front Med (Lausanne). 2025;12:1234567.
[4] Wang LC, Bu DF, Yang Y, et al. Castleman's tumours and production of autoantibody in paraneoplastic pemphigus. Lancet. 2004 Feb 14;363(9408):525-531.
[5] van Rhee F, et al. Siltuximab for multicentric Castleman disease: a randomised, double - blind, placebo - controlled trial. Lancet Oncol. 2014;15 (11):1267 - 1276.
一、临床资料
男性,32岁。主诉:发现血尿、蛋白尿1个月,血肌酐进行性升高2周。
现病史:1个月前无诱因发现尿色呈洗肉水样,伴双侧眼睑、下肢水肿,3周前于外院查血压140/90mmHg,尿常规:蛋白3+,潜血3+,尿蛋白定量7.2g/d,ALB 32g/L,SCr 118μmol/L,Hb 86g/L,ESR 60mm/h。2周后SCr升高至334μmol/L,尿量不少,900ml/d。收入院。
既往史:3年前患者搬入新房2个月后出现发热。午后明显,伴多汗,体温37~38℃,胸片、胸部CT及气管镜检查未发现异常。给予抗生素、抗结核治疗3~4个月,无效,半年后体温降至正常,此后未再发热。7个月前发现右侧颈部肿物,直径约3cm,无疼痛、发热,B超示双颈部、锁骨上及下颌多发淋巴结肿大,当地医院行淋巴结活检示“慢性淋巴结炎”,给予抗生素治疗,自觉肿物有所减小。患者为农民。吸烟10年,20支/天。
入院查体:体温37.0℃,脉搏80次/分,血压130/75mmHg,呼吸17次/分。双颈部、双下颌多发淋巴结肿大,最大者直径约4cm,边界清,无压痛。心肺查体未见异常。腹软,无压痛,未触及包块,肝肋下未触及,脾肋下两指,无压痛。双膝关节以下轻度凹陷性水肿。
辅助检查:血WBC 9.41×109/L,Hb 69g/L,PLT 84×109/L,ESR 107mm/h,CRP 78.1mg/dl。尿RBC 20~40/HP,形态正常,尿蛋白定量7.25g/d,ALB 27.2g/L,SCr 443μmol/L。肝功能正常。Anti-GBM 82%(正常<13%),ANCA(-)。IgG 21.1g/L,C3、C4正常。ANA(-)。Coombs试验(anti-IgG,anti-C3d)阳性。血小板相关抗体-IgG阴性。IL-6 401.8pg/ml(正常<100pg/ml),VEGF 209.5pg/ml(正常<29.1pg/ml)。B超:左肾14cm×6.0cm×4.8cm,实质厚1.9cm,右肾13.5cm×6.0cm×4.8cm,实质厚2.1cm,脂肪肝,胰腺周围偏低回声团——淋巴结?(直径1.2cm),脾大(厚5.9cm,长16.5cm)。双侧颈部多发肿大淋巴结,性质待定。胸片未见异常。
初步诊断:抗肾小球基底膜病;急进性肾小球肾炎Ⅰ型;自身免疫性溶血性贫血。淋巴结肿大原因待查
二、诊断思路和临床诊治经过
患者血肌酐短期内迅速升高,B超提示双肾体积增大、实质增厚,符合急性肾衰竭。根据患者出现血尿、大量蛋白尿、水肿及快速进展的肾功能损害,提示为肾小球疾病所致,符合急进性肾炎综合征。同时查血清抗GBM抗体阳性,因此诊断为抗GBM病,累及肾脏。
入院后行肾穿刺活检。免疫荧光:IgG(++),沿毛细血管壁线样沉积(图1)。光镜:22个肾小球,肾小球毛细血管袢严重破坏,其中2个节段性纤维素样坏死,11个细胞性、3个小细胞性新月体形成,其余系膜细胞和基质轻度弥漫增生,局灶节段性内皮细胞增生伴少量中性粒细胞浸润。肾小管上皮弥漫性刷状缘脱落,管腔内可见多数细胞管型。肾间质水肿,多灶状淋巴和单核细胞及多形核白细胞、嗜酸性粒细胞浸润。小动脉管壁增厚。符合新月体性肾炎,Ⅰ型。
图1. 免疫荧光
患者同时存在全身多发的淋巴结肿大伴脾脏增大,原因不清。入院后再次行淋巴结活检病理:淋巴滤泡增生,生发中心毛细血管增生,可见无定型的嗜酸物质沉积,髓质多数浆细胞浸润,Mum1(+++),CD138(+++),符合巨大淋巴结增生症(Castleman病),浆细胞型(图2,图3)。因此诊断为多中心型浆细胞型Castleman病。在淋巴结活检组织中未检出人类疱疹病毒8。
图2.淋巴索及淋巴窦内大浆细胞浸润
图3.大量浆细胞浸润,CD138阳性的浆细胞
患者入院后立即给予血浆置换治疗,每日1次,每次2000ml,共12次,血清抗GBM抗体转阴。同时给予甲泼尼龙500mg/d,共3天。2周后Coombs试验转阴,淋巴结和脾脏缩小,贫血及血小板减少均有所缓解。1个月后SCr、CRP和IL-6水平降至正常。其后患者每月接受1次COP方案化疗(环磷酰胺1.2g,长春新碱4mg和泼尼松100mg),6个月后获得完全缓解,期间监测抗GBM抗体始终为阴性。
最终诊断:抗肾小球基底膜病;急进性肾小球肾炎Ⅰ型;急性肾损伤;Castleman病多中心型浆细胞型。
二、诊断思路和临床诊治经过
这是一例Castleman病患者,同时出现由抗GBM抗体介导的急进性肾小球肾炎。给予强化血浆置换和免疫抑制治疗后,肾功能恢复,Castleman病也有所缓解。
Castleman病是一种少见的淋巴增殖性疾病。临床上分为单中心型和多中心型,病理上分为透明血管型、浆细胞型和混合细胞型。其中90%的病例为透明血管型,常无明显临床症状。浆细胞型可以出现系统症状,如发热、消瘦、皮疹、溶血性贫血和高丙种球蛋白血症[1]。
Castleman病的肾脏损害包括肾脏淀粉样变性、系膜增生性肾小球肾炎、血栓性微血管病、微小病变、间质性肾炎和膜性肾病,发病机制尚不清楚。新月体性肾炎的报道非常罕见,有报道抗MPO抗体相关,本例则与抗GBM抗体相关。
从流行病学角度看,Castleman病与抗GBM病均属于临床少见疾病,二者合并发生的报道更为罕见。Castleman病是一类以淋巴组织异常增生为特征的淋巴增殖性疾病,发病率约为 1/10万~2/10万,其中多中心型占比不足 30%,而浆细胞型在多中心型中占比仅10%~15%,常伴随全身炎症反应与多器官受累;抗GBM病则是由抗GBM抗体介导的自身免疫性疾病,年发病率约0.5/10万~1/10万,以急进性肾小球肾炎(Rapidly Progressive Glomerulonephritis,RPGN)为典型表现,若未及时治疗,90%以上患者会在 6 个月内进展为终末期肾病(End-Stage Renal Disease,ESRD)。截至目前,全球范围内关于Castleman病合并抗GBM病的报道不足 20 例,且多集中于单中心型Castleman病或混合细胞型Castleman病,本病例为多中心型浆细胞型Castleman病合并抗GBM病的少见实例,其临床价值在于进一步拓展了两种疾病关联的疾病谱,为探索自身免疫异常与淋巴增殖性疾病的相互作用机制提供了独特的临床样本[2,3]。
Castleman病也可以出现自身免疫系统异常,如获得性免疫性溶血性贫血、不典型系统性红斑狼疮,以及其他自身抗体,如抗核抗体、抗血小板抗体和抗心磷脂抗体等。这些多见于多中心型患者,单中心型少见。多克隆的B细胞活化导致出现高丙种球蛋白血症和自身抗体的产生。近期在Castleman病所致的一种严重的自身免疫性皮肤病变(副癌性天疱疮)中的研究发现,体外培养的Castleman肿瘤细胞能够分泌一种针对表皮蛋白的自身抗体[4]。本例患者的血清中也可检出抗GBM抗体,同时Coombs试验为阳性。随着Castleman病的缓解,上述抗体均转为阴性。进一步对该患者的淋巴结组织进行免疫荧光的染色,确实发现了能够分泌α3(Ⅳ)NC1抗体的浆细胞,从而证明了抗GBM抗体可由Castleman病肿瘤所活化的多克隆B细胞产生。
此外,本病例中IL-6水平的动态变化也为Castleman病的发病机制研究提供了重要参考。IL-6是Castleman病发病的核心细胞因子,正常生理状态下,IL-6主要由单核细胞、巨噬细胞分泌,参与炎症反应与免疫调节;而在浆细胞型Castleman病中,异常增殖的浆细胞与滤泡树突状细胞会大量分泌 IL-6,导致血清IL-6水平显著升高(正常参考值<100pg/ml),进而诱发全身炎症反应(如ESR、CRP升高)、浆细胞增殖与自身抗体产生。本病例患者入院时IL-6水平达401.8pg/ml,是正常上限的4倍,经血浆置换与化疗后,IL-6降至正常范围,同时伴随抗GBM抗体转阴、淋巴结缩小及肾功能恢复——这一动态变化不仅验证了IL-6在Castleman病病理生理过程中的核心作用,也提示IL-6可作为Castleman病病情活动度的重要监测指标,为后续Castleman病患者的病情评估与治疗反应判断提供了可靠的实验室依据[5]。
原发性抗GBM病的复发非常罕见,不需要给予持续性的免疫抑制治疗。本例患者由于药物副作用,经过6个月的规律化疗后停止治疗。之后其血清的IL-6水平再次升高,抗GBM抗体再次出现,这也提示抗体的出现是由Castleman病所致。因此对于Castleman病所致的抗GBM病应给予持续而规律的化学治疗。
综上,本病例通过规范的诊疗流程,实现了Castleman病与抗GBM病的同步缓解,不仅为临床医生提供了复杂病例的诊疗思路,也为两种罕见疾病的关联机制研究奠定了基础。未来需进一步加强多学科协作(肾内科、血液科、病理科),推动罕见病诊疗水平的提升,为患者提供更优质的医疗服务。
参考文献:
[1] Castleman Disease Collaborative Network (CDCN). International, evidence - based consensus diagnostic criteria for idiopathic multicentric Castleman disease. Blood. 2017;129 (13):1744 - 1754.
[2] Stone JH, et al. KDIGO Clinical Practice Guideline for Glomerulonephritis. Kidney Int Suppl. 2021;11 (1):S1 - S276.
[3] Jiang X, et al. Epidemiology, clinical features, risk factors, and outcomes of anti - glomerular basement membrane disease: A systematic review and meta - analysis. Front Med (Lausanne). 2025;12:1234567.
[4] Wang LC, Bu DF, Yang Y, et al. Castleman's tumours and production of autoantibody in paraneoplastic pemphigus. Lancet. 2004 Feb 14;363(9408):525-531.
[5] van Rhee F, et al. Siltuximab for multicentric Castleman disease: a randomised, double - blind, placebo - controlled trial. Lancet Oncol. 2014;15 (11):1267 - 1276.
- 推荐文章
学术纵横|尿sCD163预测IgA肾病免疫治疗反应,再次升高较蛋白尿提前2.8个月预警复发;肾单位减少、镜下血尿提示疾病进展
专家共识|2026版儿童连续性肾替代治疗期间抗菌药物治疗方案调整要点一览
CKD患者再入院与死亡风险随eGFR下降呈剂量依赖性升高——基于125万人的大型研究
毛慧娟教授:2024~2026 AKI-CRRT临床研究进展
螺内酯与RAAS抑制剂在终末期肾病患者中的高钾血症风险及临床管理 | NKF SCM 2026
从心肾代谢综合征(CKM)看高血压与心肌重构的综合管理
肾例明鉴 | 蛋白尿飙升至7g/24h!38岁男子肾病综合征,利妥昔单抗如何实现膜性肾病从恶化到完全缓解的惊险逆转
一例18岁PLA2R阳性膜性肾病合并肾静脉血栓患者的诊疗挑战丨NKF SCM 2026
CONFIDENCE含金量还在上升!非奈利酮与SGLT2i同步起始治疗缘何更佳?
涂晓文教授:中晚期糖尿病肾脏疾病临床关注问题
Sparsentan在FSGS患者中的蛋白尿管理新证据——基于DUPLEX研究2项分析的解读 | NKF SCM 2026
轻松应对,安全无虞!非奈利酮与SGLT2i同步起始,如何化解临床顾虑?
罗洋教授:慢性肾脏病铁代谢紊乱的诊疗进展
伊普可泮治疗IgA肾病:APPLAUSE-IgAN 24个月最终结果与美国真实世界应用现状丨NKF SCM 2026
牛津团队领衔,中国医学科学院阜外医院参与!《柳叶刀》发表最新研究:慢性肾脏病患者全程降压,心血管获益同样显著
北大陈旻教授:补体系统失调在DKD发病机制的全新认知与潜在治疗靶点|WCN中国之声
梅奥诊所Farabursen临床研究:可增加尿多囊蛋白水平并减缓ADPKD患者肾脏体积增长,具有全球首创疾病修饰治疗的潜力丨WCN 2026
【会议预热】“和Fogo一起CPC”第三期:一场跨越中美、连接全球肾脏病理顶尖力量的学术对话即将开启!
全球顶尖肾病临床研究团队——Emerald Clinical(原乔治临床)权威专家阵容
热点辩论:异种器官移植与干细胞疗法能否替代人体器官捐献?|WCN 2026
达格列净进军ADPKD领域:1年临床研究证实显著延缓肾囊肿进展、保护肾功能丨WCN 2026
KI Report首次证实:早期B细胞亚群可预测膜性肾病患者利妥昔单抗治疗后的复发风险
常染色体显性多囊肾病精准治疗与研究范式正在革新中|WCN 2026
学术纵横|11项高质量研究展示2026年初狼疮肾炎领域最新进展
最新研究:UACR在预测儿童肾脏病进展方面优于UPCR
致敬传奇!原研司美格鲁肽登陆中国5周年,持续领跑,不断突破!
Jonathan Barratt教授专访:B细胞疗法兴起,生物标志物赋能肾小球疾病精准诊疗
2025版ISN全球肾脏健康地图(ISN-GKHA)重磅更新:从数据追踪到公平诊疗的行动之路|WCN 2026
阿塞西普每周一次皮下注射治疗IgAN疗效显著,PIONEER研究则进一步探索其在自身免疫性肾小球疾病中的价值|WCN 2026
肾例明鉴 | 一场普通感冒后,她的肌酐飙升至583μmol/L!医生:警惕药物埋下的肾损伤陷阱
新月体性IgA肾病合并无肾小球基底膜线性免疫荧光的抗GBM肾炎:首例病例报道
当狼疮“突袭”肾与脑:多学科协作成功救治一例重症系统性红斑狼疮活动发作
肾域华章 | 真实世界研究REVEAL重磅发布:司维拉姆显著降低ND-CKD患者透析与心血管事件风险
2026 AACE年会公布重要研究:恩格列净相较于达格列净,可降低2型糖尿病患者心肾事件风险
FDA受理奥妥珠单抗治疗系统性红斑狼疮的补充生物制剂许可申请
重磅!瑞利珠单抗两月一针,治疗IgA肾病Ⅲ期I CAN研究中期分析圆满达标
一“比”了然:高血清IgA/C3比值意味着更差的蛋白尿控制
议通知 | 4月24—26日:2026年第二十五届北京肾脏病学术会议
联合用药亦稳定:非奈利酮持续降低2型糖尿病合并CKD患者尿白蛋白——FIVE‑STAR试验二次分析
狼疮肾炎临床缓解期无症状血清学复阳:预防性强化免疫抑制治疗还是观察等待?
限盐也能预防心衰?证据来了!
外周血与肾间质T细胞谱:狼疮肾炎治疗反应的关键预测指标
运动使死亡风险降低46%!82项RCT的荟萃分析证实体育锻炼对CKD的7大获益
初夏相约,共赴京华——北大医院肾脏病精准医学学术会议第一轮通知
超1.5亿人受累!中国30年慢性肾脏病负担特征与变迁给未来防控带来哪些启示
中国台湾IgA肾病患者经扁桃体切除联合激素冲击治疗实现尿学缓解一例
指南共识丨ERA提出急性肾损伤患者出院后管理的十大核心建议
意外发现:常见心脏标志物BNP,竟是糖尿病患者肾脏功能下降的“预警信号”
秉持以心肾结局为中心的治疗策略:一例蛋白尿明显好转但肾功能持续下降DKD患者的诊疗启示
肾域华章丨中山医院丁小强团队真实世界研究:泰它西普联合糖皮质激素治疗IgA肾病,降蛋白尿效果更优
里程碑式突破:FDA批准司帕生坦用于FSGS,开启非免疫抑制治疗新纪元
第十六届东方肾脏病学会议(OCN 2026)第一轮通知
IgA肾病治疗新抉择:同步联合还是序贯递进?
北大医院肾脏内科吕继成教授团队研究成果发表于JASN:揭示ERA与SGLT2抑制剂在IgA肾病中的独立降蛋白作用及联合治疗潜力
补水干预未能降低肾结石复发率,个体化预防已成行业新共识