
- 首页 > 正文
万建新教授:乳酸代谢异常与糖尿病肾病
发表时间:2026-05-23 17:07:55
编者按
传统观点将乳酸视为无氧代谢终末废物,现代研究证实乳酸是重要能量底物、信号分子与炎症调控因子;乳酸化作为一种新的表观遗传修饰,可调控基因转录与细胞代谢,参与肾脏疾病进程[1]。糖尿病状态下,乳酸生成与清除失衡引发的代谢异常,是驱动DKD发生发展的关键代谢环节。
在第十三届肾脏病学新进展西湖论坛上,福建医科大学附属第一医院万建新教授以“乳酸代谢异常与糖尿病肾病”为题作专题报告,系统阐述了乳酸代谢紊乱在糖尿病肾病(DKD)发生发展中的核心作用、分子机制及诊疗价值,为DKD机制研究与靶向干预提供了全新视角。
葡萄糖经一系列糖酵解酶介导的反应生成丙酮酸。正常氧条件下,丙酮酸转运至线粒体并进入三羧酸循环;缺氧环境中,乳酸脱氢酶A(LDHA)催化丙酮酸转化为乳酸。
乳酸通过4种途径代谢:①氧充足时,乳酸可经乳酸脱氢酶B(LDHB)氧化为丙酮酸,并进入线粒体参与三羧酸循环;②乳酸可通过糖异生途径转化为葡萄糖;③乳酸通过代谢中间产物乙酰辅酶A间接参与脂肪酸、胆固醇或酮体的合成;④当血乳酸水平异常升高时,乳酸可通过尿液、汗液排出[1]。肾脏作为重要乳酸代谢器官,其乳酸稳态失衡与DKD密切相关(图1)。
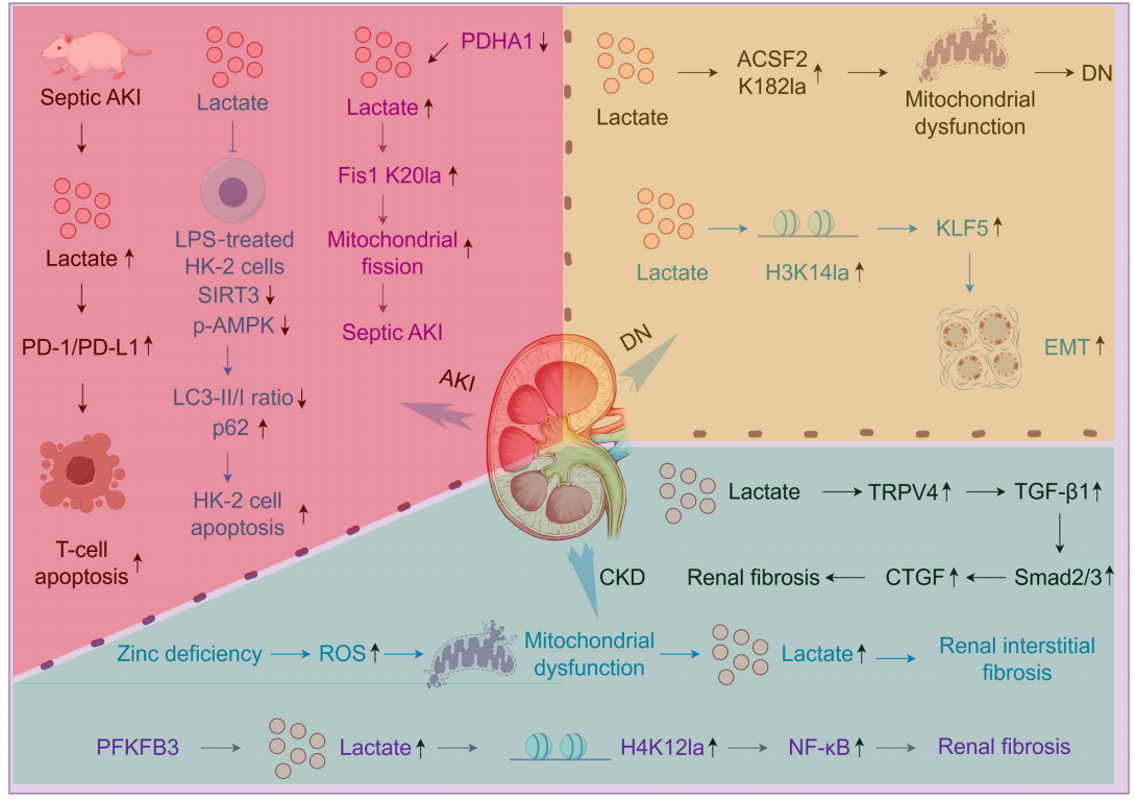
图1. 肾脏疾病中乳酸和乳酸酸化的机制[1]
二、糖尿病状态下乳酸代谢的异常
糖尿病状态下,乳酸生成增加、清除利用障碍,共同导致高乳酸血症或局部组织乳酸蓄积,构成肾脏损伤的重要代谢基础。
01、乳酸生成增加
外周组织生成增多:肌肉、脂肪等组织对葡萄糖摄取与利用障碍,导致局部相对缺氧,糖酵解增强,乳酸生成增多。
肾脏本身乳酸产生增加:DKD患者肾脏呈高代谢状态,耗氧量增加导致肾组织尤其是肾髓质相对缺氧;高血糖激活多元醇通路,消耗NAD+,细胞更依赖无氧糖酵解供能,进一步促进乳酸产生[2]。
药物与代谢因素:糖尿病状态下,胰岛素刺激的葡萄糖摄取增加了细胞内葡萄糖和糖酵解流量;双胍类药物抑制线粒体复合物Ⅰ,损害氧化磷酸化,推动能量代谢转向糖酵解,这均会增强乳酸生成[2]。
02、肝脏乳酸清除与利用障碍
肝脏代谢能力下降:糖尿病常合并非酒精性脂肪肝,肝脏糖异生功能受损,其将乳酸转化为葡萄糖的效率降低。
肾脏乳酸清除能力减退:肾脏是仅次于肝脏的乳酸清除器官,DKD进展导致肾功能下降,乳酸滤过与代谢降解能力显著降低,加剧体内蓄积[2]。
三、乳酸蓄积对肾脏的损伤
乳酸蓄积通过促进肾脏纤维化、介导炎症反应、诱发代谢重构和线粒体功能障碍及表观遗传修饰异常,多维度损伤肾脏,推动DKD的发生与进展(图2)。
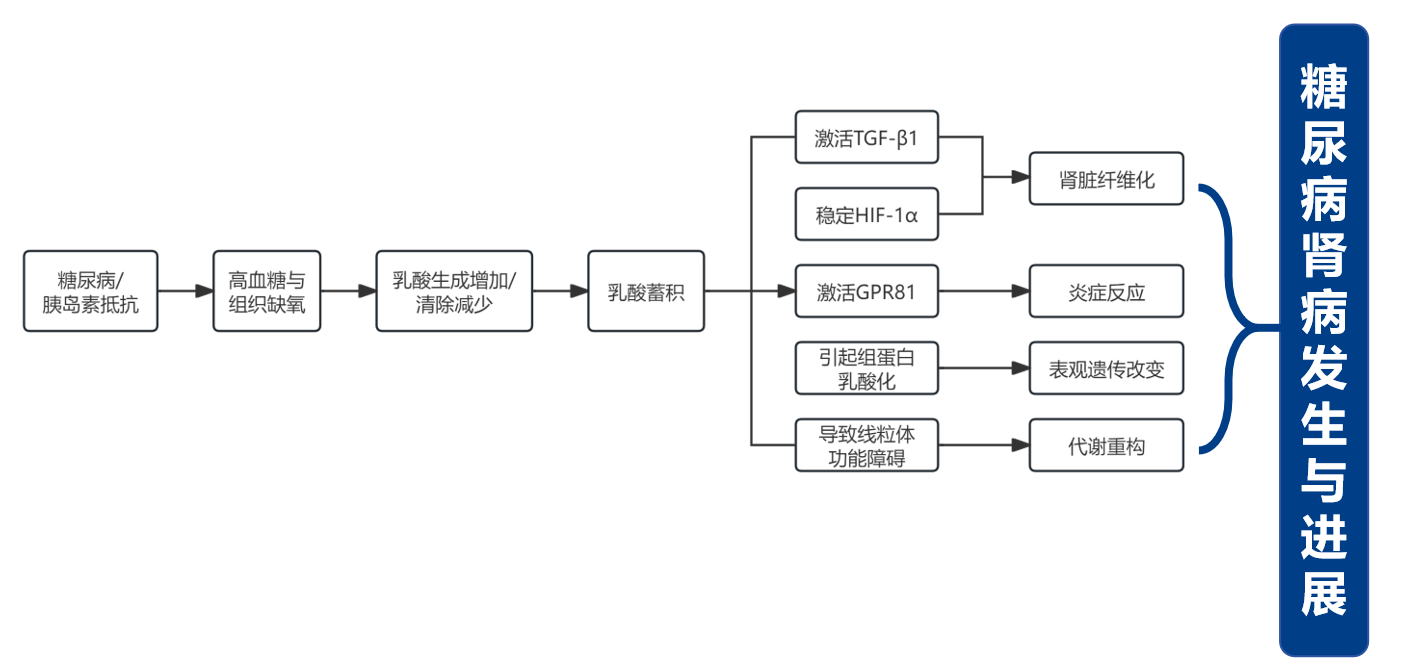
图2. 乳酸蓄积对肾脏损伤的损伤途径
01、促进肾脏纤维化
激活转化生长因子-β1(TGF-β1):乳酸直接刺激肾小管上皮细胞与成纤维细胞表达TGF-β1,诱导细胞外基质大量沉积,引发肾间质纤维化[3]。
诱导上皮间质转化(EMT):乳酸联合TGF-β1,促使肾小管上皮细胞极性丧失,转化为肌成纤维细胞,加速纤维化进程[3]。
稳定缺氧诱导因子-1α(HIF-1α):乳酸抑制HIF-1α降解,其持续激活促进促纤维化基因表达,放大纤维化效应[3]。
02、介导免疫调节异常及加剧炎症反应
乳酸通过单羧酸转运蛋白(MCTs)调控细胞内水平,细胞外乳酸激活G蛋白偶联受体GPR81,抑制cAMP及下游促炎通路,同时乳酸与葡萄糖、脂肪酸衍生的代谢物竞争进入线粒体代谢[2]。而作为一种表观遗传调控因子,细胞内乳酸可为组蛋白酰化提供酰基。
乳酸是GPR81的天然配体,其激活GPR81后,促进肾脏固有细胞分泌MCP-1、IL-6等炎症因子,招募巨噬细胞等炎症细胞浸润肾组织,形成慢性炎症微环境,破坏肾单位结构[4]。
04、导致代谢重构&线粒体功能障碍
蓄积的乳酸可导致能量代谢紊乱,其抑制线粒体的氧化磷酸化,使细胞更加依赖低效糖酵解供能,形成“缺氧-糖酵解增强-乳酸蓄积”恶性循环。当前认为,线粒体内共生的中断是炎症性疾病的基础,线粒体核酸可能通过诱导细胞因子成为免疫反应的关键触发器[5,6]。
05、乳酸化修饰介导肾脏损伤
蛋白质乳酸化是一种蛋白质翻译后的修饰。赖氨酸乳酸化是通过在赖氨酸残基上添加乳酰基来调节基因表达、代谢和细胞信号传导,在肾脏病的发生和发展中起重要作用[7]。
H3K14la-KLF5轴与DKD肾小管EMT和纤维化:EMT是肾小管间质纤维化的重要环节。高糖导致糖酵解增强、乳酸升高,乳酸进一步增加H3K14la。H3K14la作为组蛋白修饰,增强KLF5表达,KLF5再调控EMT相关基因,最终促进肾小管纤维化表型[3]。
PFKFB3-H4K12la-NF-κB轴与纤维化:PFKFB3是糖酵解增强的重要调控酶,其上调促进乳酸积累,增强H4K12乳酸化,而H4K12la促进NF-κB家族相关基因激活;抑制PFKFB3可降低肾乳酸水平,减轻炎症和纤维化[8]。
ACSF2-K182乳酸化与肾小管线粒体损伤:高糖诱导糖酵解增强,乳酸积累,肾小管蛋白赖氨酸乳酸化升高,增强ACSF2 K182位点乳酰化,从而诱导线粒体功能障碍,加速肾小管损伤与DKD进展[9]。
TRIM65-K206乳酸化与糖酵解/铁死亡:DKD高乳酸环境下,乳酸可促进TRIM65 K206位点乳酸化,降低其E3泛素连接酶活性,使其不能有效降解IREB2和PDK4,铁死亡和糖酵解同时增强,最终促进肾小管损伤[10]。
LARS1-K970乳酸化与足细胞自噬障碍:高糖诱导足细胞乳酸积累并促进蛋白质乳酸化,其中LARS1 K970乳酰化增强,激活mTORC1通路,抑制自噬,导致足细胞损伤与蛋白尿[11]。
乳酸蓄积通过多靶点、多通路对多种功能蛋白的乳酸化修饰,形成表观遗传调控网络,持续推动DKD进展。
四、乳酸作为评估DKD的潜在生物标志物
乳酸水平可作为 DKD 早期诊断、病情评估与预后判断的潜在生物标志物,具备临床转化价值。
研究证实,尿乳酸水平与白蛋白尿、肾小管损伤及上皮应激标志物显著相关,可独立预测2型糖尿病患者终末期肾病(ESRD)发生风险[12];尿乳酸、血乳酸水平、尿乳酸/肌酐比值与DKD严重程度及进展速度呈正相关,有望成为便捷的预后评估指标[12]。
抑制乳酸生成:研究表明,SGLT2抑制剂(可降低肾小球高滤过和高代谢,改善缺氧)可能部分通过纠正乳酸代谢失衡发挥肾脏保护作用;
靶向乳酸信号通路:GPR81拮抗剂有望阻断乳酸的致炎和促纤维化信号;干预组蛋白乳酸化,探索特异性抑制剂以逆转异常的基因表达;
改善肾脏微循环与氧供:旨在从根源上缓解组织缺氧。
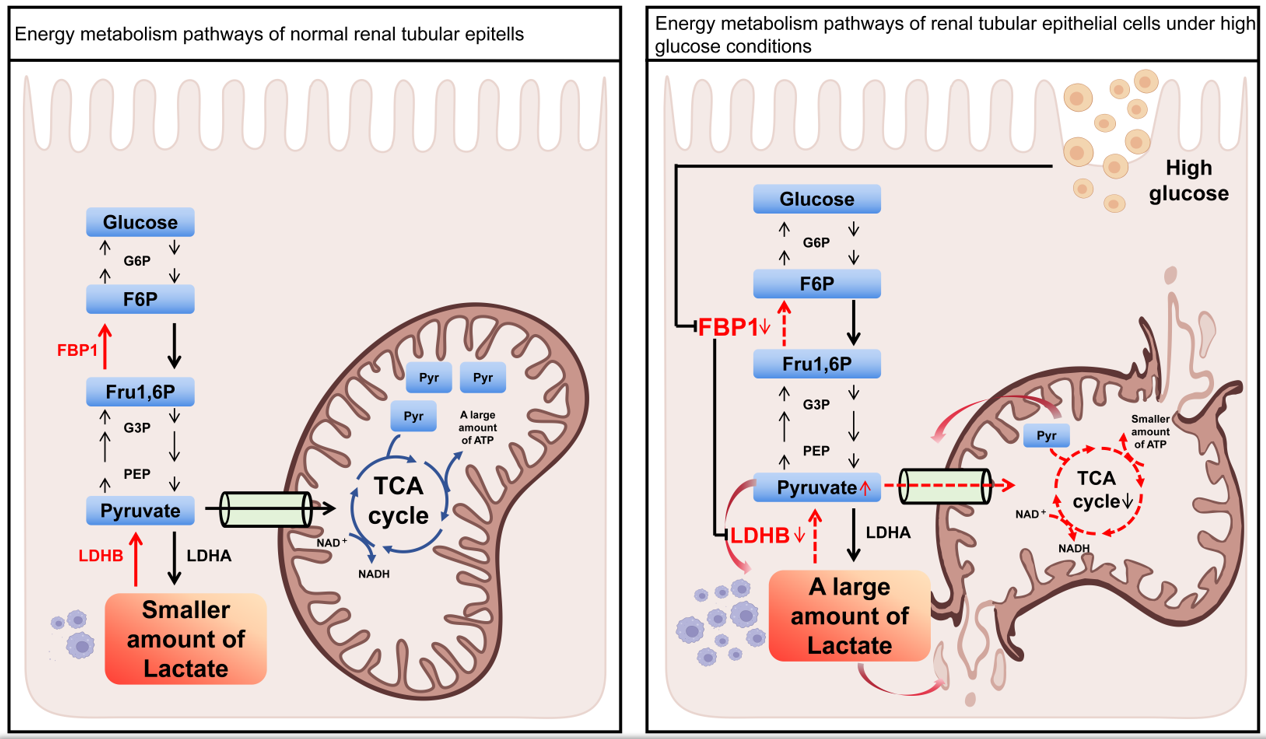
图3. 正常肾小管上皮细胞(左图)和高糖条件下肾小管上皮细胞(右图)的能量代谢通路(研究尚待发表)
五、总结
乳酸代谢异常是连接糖尿病代谢紊乱与肾脏损伤的核心桥梁。乳酸从传统代谢废物转变为驱动DKD进展的关键致病因子,通过促进纤维化、加剧炎症、诱导线粒体损伤及表观遗传修饰异常,多维度介导肾损伤。尿乳酸、血乳酸等可作为DKD潜在生物标志物;靶向乳酸生成、信号通路及代谢循环的干预策略,有望打破病理恶性循环,为DKD防治提供全新方向。
专家简介

肾内科主任
科主任、主任医师、教授、博士生导师
中国医师协会肾脏内科医师分会常委
中国医院协会血液净化中心分会常委
中国研究型医院学会肾脏病专委会常委
中华医学会肾脏病学分会第十届常委
福建省医学会肾脏病学分会第四、五届主任委员
[1] Int J Mol Med. 2025 Aug;56(2):121.
[2] Cells, 2025, 14(14): 1096.
[3] Redox Biol, 2024, 75: 103246.
[4] Cell Death Dis, 2022, 13(4): 340.
[5] Nature. 2024 Feb;626(7998):271-279.
[6] JCI Insight. 2024;9(11):e168825.
[7] Front Cell Dev Biol. 2025;13:1533175.
[8] Kidney Int, 2024, 106(2): 226-240.
[9] Diabetologia, 2024, 67(7): 1429-1443.
[10] Cell Rep, 2025, 44(8): 116091.
[11] Cell Signal, 2025, 134: 111955.
[12] Kidney Int, 2023, 104(6): 1135-1149.
[13] Ren Fail, 2025, 47(1): 2469746.
传统观点将乳酸视为无氧代谢终末废物,现代研究证实乳酸是重要能量底物、信号分子与炎症调控因子;乳酸化作为一种新的表观遗传修饰,可调控基因转录与细胞代谢,参与肾脏疾病进程[1]。糖尿病状态下,乳酸生成与清除失衡引发的代谢异常,是驱动DKD发生发展的关键代谢环节。
在第十三届肾脏病学新进展西湖论坛上,福建医科大学附属第一医院万建新教授以“乳酸代谢异常与糖尿病肾病”为题作专题报告,系统阐述了乳酸代谢紊乱在糖尿病肾病(DKD)发生发展中的核心作用、分子机制及诊疗价值,为DKD机制研究与靶向干预提供了全新视角。

葡萄糖经一系列糖酵解酶介导的反应生成丙酮酸。正常氧条件下,丙酮酸转运至线粒体并进入三羧酸循环;缺氧环境中,乳酸脱氢酶A(LDHA)催化丙酮酸转化为乳酸。
乳酸通过4种途径代谢:①氧充足时,乳酸可经乳酸脱氢酶B(LDHB)氧化为丙酮酸,并进入线粒体参与三羧酸循环;②乳酸可通过糖异生途径转化为葡萄糖;③乳酸通过代谢中间产物乙酰辅酶A间接参与脂肪酸、胆固醇或酮体的合成;④当血乳酸水平异常升高时,乳酸可通过尿液、汗液排出[1]。肾脏作为重要乳酸代谢器官,其乳酸稳态失衡与DKD密切相关(图1)。
图1. 肾脏疾病中乳酸和乳酸酸化的机制[1]
二、糖尿病状态下乳酸代谢的异常
糖尿病状态下,乳酸生成增加、清除利用障碍,共同导致高乳酸血症或局部组织乳酸蓄积,构成肾脏损伤的重要代谢基础。
01、乳酸生成增加
外周组织生成增多:肌肉、脂肪等组织对葡萄糖摄取与利用障碍,导致局部相对缺氧,糖酵解增强,乳酸生成增多。
肾脏本身乳酸产生增加:DKD患者肾脏呈高代谢状态,耗氧量增加导致肾组织尤其是肾髓质相对缺氧;高血糖激活多元醇通路,消耗NAD+,细胞更依赖无氧糖酵解供能,进一步促进乳酸产生[2]。
药物与代谢因素:糖尿病状态下,胰岛素刺激的葡萄糖摄取增加了细胞内葡萄糖和糖酵解流量;双胍类药物抑制线粒体复合物Ⅰ,损害氧化磷酸化,推动能量代谢转向糖酵解,这均会增强乳酸生成[2]。
02、肝脏乳酸清除与利用障碍
肝脏代谢能力下降:糖尿病常合并非酒精性脂肪肝,肝脏糖异生功能受损,其将乳酸转化为葡萄糖的效率降低。
肾脏乳酸清除能力减退:肾脏是仅次于肝脏的乳酸清除器官,DKD进展导致肾功能下降,乳酸滤过与代谢降解能力显著降低,加剧体内蓄积[2]。
三、乳酸蓄积对肾脏的损伤
乳酸蓄积通过促进肾脏纤维化、介导炎症反应、诱发代谢重构和线粒体功能障碍及表观遗传修饰异常,多维度损伤肾脏,推动DKD的发生与进展(图2)。
图2. 乳酸蓄积对肾脏损伤的损伤途径
01、促进肾脏纤维化
激活转化生长因子-β1(TGF-β1):乳酸直接刺激肾小管上皮细胞与成纤维细胞表达TGF-β1,诱导细胞外基质大量沉积,引发肾间质纤维化[3]。
诱导上皮间质转化(EMT):乳酸联合TGF-β1,促使肾小管上皮细胞极性丧失,转化为肌成纤维细胞,加速纤维化进程[3]。
稳定缺氧诱导因子-1α(HIF-1α):乳酸抑制HIF-1α降解,其持续激活促进促纤维化基因表达,放大纤维化效应[3]。
02、介导免疫调节异常及加剧炎症反应
乳酸通过单羧酸转运蛋白(MCTs)调控细胞内水平,细胞外乳酸激活G蛋白偶联受体GPR81,抑制cAMP及下游促炎通路,同时乳酸与葡萄糖、脂肪酸衍生的代谢物竞争进入线粒体代谢[2]。而作为一种表观遗传调控因子,细胞内乳酸可为组蛋白酰化提供酰基。
乳酸是GPR81的天然配体,其激活GPR81后,促进肾脏固有细胞分泌MCP-1、IL-6等炎症因子,招募巨噬细胞等炎症细胞浸润肾组织,形成慢性炎症微环境,破坏肾单位结构[4]。
03、促进血管功能障碍
04、导致代谢重构&线粒体功能障碍
蓄积的乳酸可导致能量代谢紊乱,其抑制线粒体的氧化磷酸化,使细胞更加依赖低效糖酵解供能,形成“缺氧-糖酵解增强-乳酸蓄积”恶性循环。当前认为,线粒体内共生的中断是炎症性疾病的基础,线粒体核酸可能通过诱导细胞因子成为免疫反应的关键触发器[5,6]。
05、乳酸化修饰介导肾脏损伤
蛋白质乳酸化是一种蛋白质翻译后的修饰。赖氨酸乳酸化是通过在赖氨酸残基上添加乳酰基来调节基因表达、代谢和细胞信号传导,在肾脏病的发生和发展中起重要作用[7]。
H3K14la-KLF5轴与DKD肾小管EMT和纤维化:EMT是肾小管间质纤维化的重要环节。高糖导致糖酵解增强、乳酸升高,乳酸进一步增加H3K14la。H3K14la作为组蛋白修饰,增强KLF5表达,KLF5再调控EMT相关基因,最终促进肾小管纤维化表型[3]。
PFKFB3-H4K12la-NF-κB轴与纤维化:PFKFB3是糖酵解增强的重要调控酶,其上调促进乳酸积累,增强H4K12乳酸化,而H4K12la促进NF-κB家族相关基因激活;抑制PFKFB3可降低肾乳酸水平,减轻炎症和纤维化[8]。
ACSF2-K182乳酸化与肾小管线粒体损伤:高糖诱导糖酵解增强,乳酸积累,肾小管蛋白赖氨酸乳酸化升高,增强ACSF2 K182位点乳酰化,从而诱导线粒体功能障碍,加速肾小管损伤与DKD进展[9]。
TRIM65-K206乳酸化与糖酵解/铁死亡:DKD高乳酸环境下,乳酸可促进TRIM65 K206位点乳酸化,降低其E3泛素连接酶活性,使其不能有效降解IREB2和PDK4,铁死亡和糖酵解同时增强,最终促进肾小管损伤[10]。
LARS1-K970乳酸化与足细胞自噬障碍:高糖诱导足细胞乳酸积累并促进蛋白质乳酸化,其中LARS1 K970乳酰化增强,激活mTORC1通路,抑制自噬,导致足细胞损伤与蛋白尿[11]。
乳酸蓄积通过多靶点、多通路对多种功能蛋白的乳酸化修饰,形成表观遗传调控网络,持续推动DKD进展。
四、乳酸作为评估DKD的潜在生物标志物
乳酸水平可作为 DKD 早期诊断、病情评估与预后判断的潜在生物标志物,具备临床转化价值。
研究证实,尿乳酸水平与白蛋白尿、肾小管损伤及上皮应激标志物显著相关,可独立预测2型糖尿病患者终末期肾病(ESRD)发生风险[12];尿乳酸、血乳酸水平、尿乳酸/肌酐比值与DKD严重程度及进展速度呈正相关,有望成为便捷的预后评估指标[12]。
五、基于乳酸代谢异常的DKD治疗策略
抑制乳酸生成:研究表明,SGLT2抑制剂(可降低肾小球高滤过和高代谢,改善缺氧)可能部分通过纠正乳酸代谢失衡发挥肾脏保护作用;
靶向乳酸信号通路:GPR81拮抗剂有望阻断乳酸的致炎和促纤维化信号;干预组蛋白乳酸化,探索特异性抑制剂以逆转异常的基因表达;
改善肾脏微循环与氧供:旨在从根源上缓解组织缺氧。
六、中国研究:乳酸代谢异常在DKD进展的作用
图3. 正常肾小管上皮细胞(左图)和高糖条件下肾小管上皮细胞(右图)的能量代谢通路(研究尚待发表)
五、总结
乳酸代谢异常是连接糖尿病代谢紊乱与肾脏损伤的核心桥梁。乳酸从传统代谢废物转变为驱动DKD进展的关键致病因子,通过促进纤维化、加剧炎症、诱导线粒体损伤及表观遗传修饰异常,多维度介导肾损伤。尿乳酸、血乳酸等可作为DKD潜在生物标志物;靶向乳酸生成、信号通路及代谢循环的干预策略,有望打破病理恶性循环,为DKD防治提供全新方向。
专家简介
肾内科主任
科主任、主任医师、教授、博士生导师
中国医师协会肾脏内科医师分会常委
中国医院协会血液净化中心分会常委
中国研究型医院学会肾脏病专委会常委
中华医学会肾脏病学分会第十届常委
福建省医学会肾脏病学分会第四、五届主任委员
福建省医师协会肾脏内科医师分会首任会长
[1] Int J Mol Med. 2025 Aug;56(2):121.
[2] Cells, 2025, 14(14): 1096.
[3] Redox Biol, 2024, 75: 103246.
[4] Cell Death Dis, 2022, 13(4): 340.
[5] Nature. 2024 Feb;626(7998):271-279.
[6] JCI Insight. 2024;9(11):e168825.
[7] Front Cell Dev Biol. 2025;13:1533175.
[8] Kidney Int, 2024, 106(2): 226-240.
[9] Diabetologia, 2024, 67(7): 1429-1443.
[10] Cell Rep, 2025, 44(8): 116091.
[11] Cell Signal, 2025, 134: 111955.
[12] Kidney Int, 2023, 104(6): 1135-1149.
[13] Ren Fail, 2025, 47(1): 2469746.
- 推荐文章
CCBPC 2026|马坤岭教授:肠道菌群介导DKD肾损伤机制及转化应用新进展
北大医院近期科研新发现,速览!
【会议预热】三地联动,大咖云集:肾英无界・中外IgA肾病学术前沿对话即将启幕
聚焦重症肾脏病前沿 交流血液净化新进展——中华医学会肾脏病学分会第二十届重症肾脏病与血液净化大会在合肥开幕
两例罕见病例:FSGS顶端变异型合并IgA肾病及肾内B细胞浸润1例,产后IgA肾病与TMA的重叠1例
汪年松教授:2026年ADA糖尿病肾脏病指南核心解读
重新审视CKD尿蛋白检测——迈向白蛋白尿评估的全球共识
IgA肾病“双联治疗”真实世界结果:双重内皮素-血管紧张素受体拮抗剂联合靶向释放布地奈德,蛋白尿下降、eGFR趋稳
赵建荣教授:新时期慢性肾脏病的防控与设计
破译黏膜免疫“指挥棒”:浆细胞样树突状细胞通过TLR9-APRIL轴驱动IgA肾病的关键致病分子
狼疮足细胞病激素依赖、频繁复发:传统激素节约方案失败,奥妥珠单抗能否破局?
万建新教授:乳酸代谢异常与糖尿病肾病
揭秘心肾共病密码!miRNAs既可致心脏损伤,又能预警肾病进展
全球首例病例报道!甘草诱发罕见急性间质性肾炎一例
周晓霜教授:人工智能——从眼睛看肾脏
IgAN进入“机制优先时代”——重塑诊断、治疗与缓解的临床新格局
替那帕诺可降低透析高磷血症慢性肾脏病患者血磷水平且不影响其他血清电解质
北京大学第一医院杨莉教授团队原创AI体系开启肾病智能诊疗新时代
全球原发性肾小球肾炎流行病学、临床特征及预后解析
登顶《新英格兰医学杂志》!北大医院肾脏内科张宏/吕继成教授团队开创IgA肾病治疗“中国方案”
肾例明鉴|婴幼儿多饮多尿需警惕,不及时诊治可致死,不可忽视的婴幼儿肾小管罕见病
直击源头、AI赋能——锚定CKM早期,筑牢全生命周期防线
群贤毕至绍兴城,双会合璧启新章——第六届全球华人肾脏病学术大会暨第十三届肾脏病学新进展西湖论坛正式开幕
肾识百科|急性肾损伤患者透析:只是临时过渡,还是终身宿命?
颠覆常识!《柳叶刀》发布美国尿石症研究联盟最新试验:多喝水难防结石复发,个体化预防成新趋势
日程上线 欢迎查询 | 中华医学会肾脏病学分会第二十届重症肾脏病与血液净化大会
超越KDIGO指南,JAMA重磅综述3组数据+1张表格+3张图吃透IgA肾病临床核心要点与前沿治疗
突破认知!肠道微生物或从根本上改变狼疮肾病的诊断与治疗格局
开幕倒计时7天 | 中华医学会肾脏病学分会第二十届重症肾脏病与血液净化大会
学术纵横|2026年膜性肾病研究前沿:中国学者多项关键研究,涵盖疾病解析、疗效预测与治疗优化
年龄绝非只是数字:CureGN队列揭示肾小球疾病全年龄段的临床轨迹
ESC-HFA 2026重磅速递:CKM领域前沿进展抢先看
学术纵横|尿sCD163预测IgA肾病免疫治疗反应,再次升高较蛋白尿提前2.8个月预警复发;肾单位减少、镜下血尿提示疾病进展
专家共识|2026版儿童连续性肾替代治疗期间抗菌药物治疗方案调整要点一览
CKD患者再入院与死亡风险随eGFR下降呈剂量依赖性升高——基于125万人的大型研究
毛慧娟教授:2024~2026 AKI-CRRT临床研究进展
螺内酯与RAAS抑制剂在终末期肾病患者中的高钾血症风险及临床管理 | NKF SCM 2026
从心肾代谢综合征(CKM)看高血压与心肌重构的综合管理
肾例明鉴 | 蛋白尿飙升至7g/24h!38岁男子肾病综合征,利妥昔单抗如何实现膜性肾病从恶化到完全缓解的惊险逆转
一例18岁PLA2R阳性膜性肾病合并肾静脉血栓患者的诊疗挑战丨NKF SCM 2026
CONFIDENCE含金量还在上升!非奈利酮与SGLT2i同步起始治疗缘何更佳?
涂晓文教授:中晚期糖尿病肾脏疾病临床关注问题
Sparsentan在FSGS患者中的蛋白尿管理新证据——基于DUPLEX研究2项分析的解读 | NKF SCM 2026
轻松应对,安全无虞!非奈利酮与SGLT2i同步起始,如何化解临床顾虑?
罗洋教授:慢性肾脏病铁代谢紊乱的诊疗进展
伊普可泮治疗IgA肾病:APPLAUSE-IgAN 24个月最终结果与美国真实世界应用现状丨NKF SCM 2026
牛津团队领衔,中国医学科学院阜外医院参与!《柳叶刀》发表最新研究:慢性肾脏病患者全程降压,心血管获益同样显著
北大陈旻教授:补体系统失调在DKD发病机制的全新认知与潜在治疗靶点|WCN中国之声
梅奥诊所Farabursen临床研究:可增加尿多囊蛋白水平并减缓ADPKD患者肾脏体积增长,具有全球首创疾病修饰治疗的潜力丨WCN 2026
【会议预热】“和Fogo一起CPC”第三期:一场跨越中美、连接全球肾脏病理顶尖力量的学术对话即将开启!
全球顶尖肾病临床研究团队——Emerald Clinical(原乔治临床)权威专家阵容
热点辩论:异种器官移植与干细胞疗法能否替代人体器官捐献?|WCN 2026
达格列净进军ADPKD领域:1年临床研究证实显著延缓肾囊肿进展、保护肾功能丨WCN 2026
KI Report首次证实:早期B细胞亚群可预测膜性肾病患者利妥昔单抗治疗后的复发风险
常染色体显性多囊肾病精准治疗与研究范式正在革新中|WCN 2026