
- 首页 > 正文
Jürgen Floege教授最新述评|超越半乳糖缺陷型IgA1:重新审视IgA2在IgA肾病中的致病驱动作用
发表时间:2026-07-07 08:48:55
摘要
IgA肾病既往发病机制研究长期以半乳糖缺陷型IgA1(Gd-IgA1)为核心,形成经典“四重打击”学说,IgA2被视作无病理作用的被动共沉积分子。但越来越多临床观察、动物实验与临床试验暴露出传统模型的固有缺陷,仅靠Gd-IgA1无法完整解释IgA肾病发病与进展。近期系列研究证实,IgA2广泛沉积于IgA肾病患者肾小球系膜区,通过激活补体、招募炎性细胞、加重肾小管间质纤维化放大肾脏损伤,完善了IgA肾病发病机制框架。本文梳理传统学说的矛盾,对比IgA1与IgA2结构及免疫功能差异,阐述IgA2致病证据,介绍整合两种IgA亚型的新型发病模型,并探讨该理论对精准治疗的指导价值。
一、传统IgA肾病“四重打击”模型的局限性
经典“四重打击”理论将Gd-IgA1作为IgA肾病核心致病分子:IgA1铰链区糖基化异常,生成具备自身抗原属性的Gd-IgA1;机体随之产生抗聚糖抗体,二者结合形成循环免疫复合物,沉积于肾小球系膜区,最终诱发肾小球炎症损伤。该理论能够解释患者肾小球IgA1沉积、血清Gd-IgA1升高两大特征,长期指导领域内基础与临床研究。
但近年多项证据证明,单纯Gd-IgA1不足以诱发IgA肾病。其一,血清Gd-IgA1升高无疾病特异性,健康人群、患者无症状亲属均可出现该指标上升;体外实验中单体Gd-IgA1无法诱导系膜细胞损伤。其二,基因编辑人源化小鼠模型中,B细胞特异性高表达Gd-IgA1,却未出现肾小球IgA沉积与肾脏病变。其三,felzartamabⅡ期临床试验显示,药物可长期改善蛋白尿,但停药后血清Gd-IgA1迅速恢复基线,肾脏保护效应持续存在,说明免疫复合物沉积才是肾脏损伤关键,而非Gd-IgA1本身。
供肾病理研究进一步佐证模型缺陷:5%~15%西方人群、40%东亚供肾存在无症状系膜IgA沉积,且沉积物仅含IgA1,无任何肾脏病理损伤;同时部分动物实验可脱离Gd-IgA1通路诱导类IgA肾病病变。上述证据表明,IgA肾病发病存在IgA1以外的关键调控因子,IgA2的病理作用亟待重新评估。
二、IgA1与IgA2结构、糖基化及免疫功能差异
人体IgA分为IgA1、IgA2两种亚型,二者铰链区结构、黏膜分布、糖基化修饰与促炎能力存在本质区别,直接决定其在肾病中截然不同的病理角色。
分布层面,血清IgA以IgA1为主(80%~90%);IgA2集中分布于菌群密集、蛋白酶丰富的肠道黏膜;上呼吸道、上消化道黏膜IgA1占优(65%~70%),结肠黏膜IgA2占比达65%,是IgA2主要合成位点。黏膜B细胞产生多聚IgA,经上皮转运形成分泌型IgA,分泌型IgA2耐受强酸与蛋白酶降解,稳定调控肠道菌群、中和致病菌毒素,构成肠道黏膜防御屏障。
结构差异集中于铰链区:IgA1铰链区长且柔性,富含O-糖基化位点,抗原识别范围广,但易被细菌蛋白酶水解;IgA2铰链区短,缺失O-糖基化位点,分子刚性强,抗蛋白酶切割,适配肠道恶劣环境。糖基化修饰调控炎症活性:IgA1唾液酸化水平高,循环单体IgA1整体呈抗炎效应;IgA2唾液酸化程度低,聚集状态下可强烈刺激巨噬细胞、中性粒细胞释放促炎因子。人为去除IgA1唾液酸后,其促炎活性接近IgA2,证明糖基化是亚型免疫病理活性的核心调控因素。
补体激活能力是二者最关键的病理分界点。补体旁路、凝集素通路激活是IgA肾病肾损伤核心驱动力,且两种亚型激活效率差异显著。单体IgA1通过CD89受体传递抑制性炎症信号;IgA2促炎效应不依赖经典CD89通路,多聚体IgA2可高效激活补体旁路与凝集素通路,诱导中性粒细胞胞外诱捕网与大量炎症因子释放。过敏性紫癜肾炎活检可见IgA1、IgA2共沉积,IgA2与凝集素通路激活、不良预后直接相关,侧面证实其促损伤作用。
三、IgA2具备独立致病放大效应的循证证据
早期研究受抗体交叉反应等技术限制,难以稳定检出肾小球IgA2,长期将其归为无意义共沉积产物。2026年Li等人发表的多中心临床与转基因小鼠研究,提供了IgA2主动介导肾损伤的直接因果证据。
临床研究纳入161例中国多中心IgA肾病肾活检标本,免疫荧光显示98%以上患者系膜区存在IgA2沉积,包含IgA2(m1)、IgA2(m2)两种同种异型,前者多见于白种人,后者为亚洲、非洲人群优势亚型。质谱定量显示肾小球总沉积IgA仍以IgA1为主,IgA2含量仅为IgA1的1/50,但具备明确病理价值;IgA肾病患者血浆IgA2较健康对照升高约30%。肾小球IgA2沉积荧光强度与病理损伤程度显著正相关:IgA2高沉积患者系膜增生、肾小管萎缩间质纤维化更严重,肾脏C3激活水平升高,估算肾小球滤过率更低,证明IgA2沉积直接推动肾脏慢性病变。
为建立因果关系,研究构建人源化IgA1、IgA2特异性转基因小鼠,经干酪乳杆菌细胞壁提取物刺激模拟黏膜免疫紊乱。结果显示,IgA2转基因小鼠补体活化、巨噬细胞浸润、肾小管萎缩程度均显著高于IgA1小鼠,证实IgA2并非被动随IgA1沉积,而是主动推动肾脏炎症与纤维化。该研究存在一定局限:临床队列仅覆盖中国人群,结论需多种族验证;人源化小鼠无法完全复刻人类自发性IgA肾病病程,但仍是目前IgA2致病最核心体内证据。
综上,IgA肾病“四重打击”发病模型需进一步更新完善。Gd-IgA1依旧是疾病核心,但现有证据证明IgA2同样发挥关键作用。肠道来源IgA2可进入血液循环,形成免疫复合物,与含IgA1的复合物共同沉积于肾小球系膜区。IgA2沉积机制、免疫复合物组分仍需借助新技术深入解析。图1展示了含IgA2循环免疫复合物的潜在作用模式:IgA2沉积后,可强效激活补体、招募巨噬细胞,加速肾小球与肾小管间质损伤。后续仍需探索IgA2免疫复合物的形成过程、抗原组分;IgA2可能通过黏膜移位大量入血,或由骨髓、淋巴组织异常过量合成。因此IgA肾病不只是IgA1糖基化异常、抗Gd-IgA1抗体生成类疾病,黏膜IgA亚型失衡同样会驱动肾脏进行性损伤。
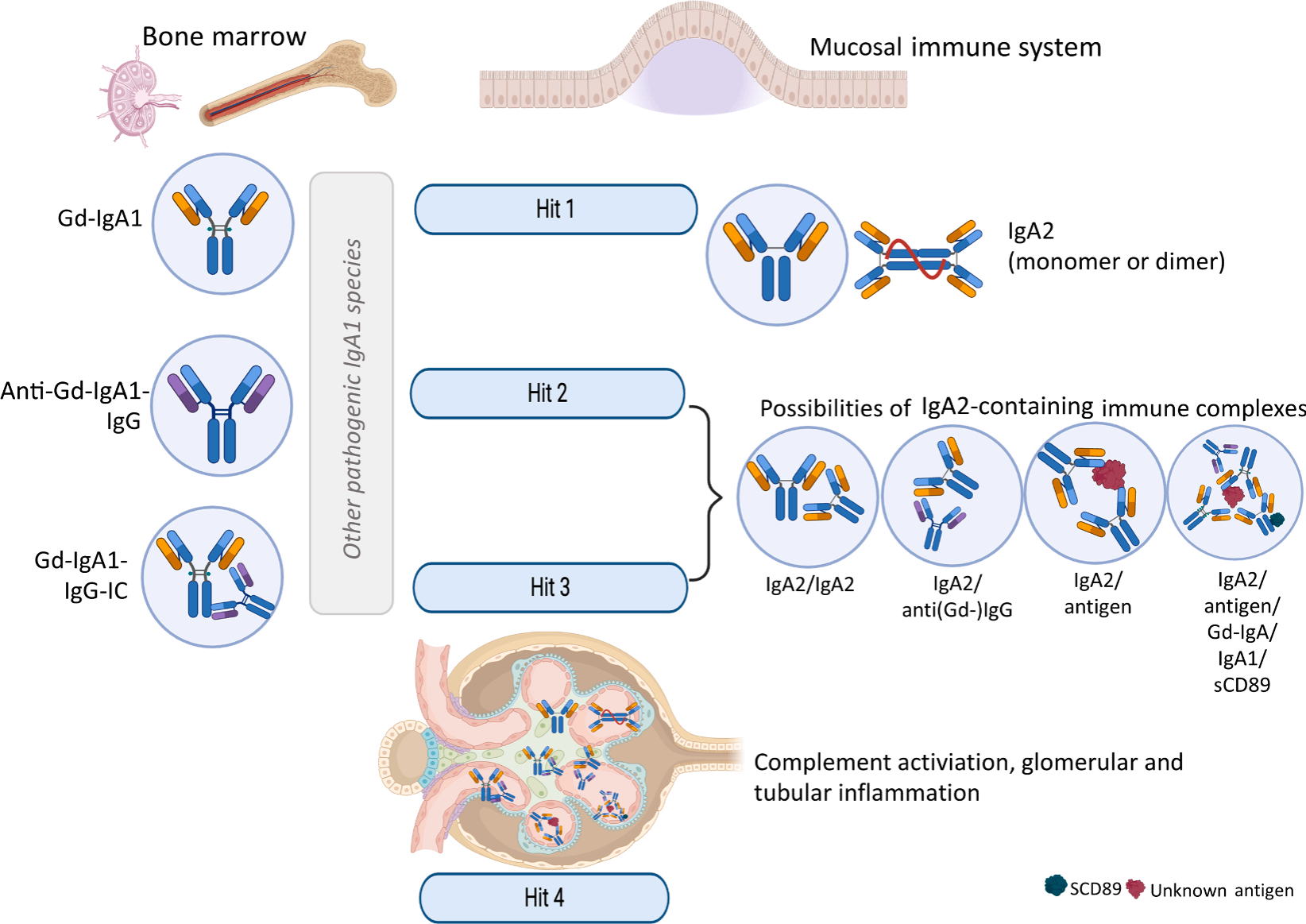
图1.IgA1和IgA2对IgA肾病贡献的整合模型
四、IgA2致病理论对临床治疗的指导价值
现有IgA肾病治疗多广谱抑制B细胞或阻断下游炎症,IgA2致病机制的明确,为分层精准治疗提供四类新思路。
第一,调控黏膜免疫与肠道菌群的疗法,可优先减少IgA2合成;
第二,靶向补体的药物,尤其是作用于凝集素、旁路通路的制剂,可特异性缓解IgA2介导的肾损伤;
第三,开发Fcα受体靶向药物,可调控IgA1、IgA2与髓系细胞的相互作用;
第四,检测IgA亚型可优化风险分层,精准识别IgA2炎症主导、进展风险更高的患者。
该理论并未否定“四重打击”学说,而是补充完善原有发病框架:IgA1启动免疫复合物生成,IgA2则调控IgA肾病炎症与纤维化进展。重新认识IgA2不会削弱Gd-IgA1的核心地位,反而将黏膜免疫、补体激活、亚型特异性炎症整合为一套更完整的发病机制。随着治疗手段不断发展,充分理解IgA1、IgA2的双重作用,是实现从单纯抑制症状到真正逆转病程、精准靶向致病IgA亚型的关键。
五、总结与展望
长久以来IgA肾病研究局限于IgA1单一亚型范式,IgA2独特的分子结构、黏膜分布与强促炎促纤维化能力,使其从“无关旁观者”转变为重要致病放大器。多中心活检证实绝大多数IgA肾病存在肾小球IgA2沉积,沉积水平与病理损伤、肾功能预后直接相关;人源化动物模型确立IgA2加重肾损伤的因果关系,搭建起IgA1、IgA2协同致病的整合模型。该理论打通黏膜免疫、IgA亚型生物学、补体激活与肾脏纤维化的关联,开辟多条全新治疗靶点。
参考文献:Seikrit C, Floege J. Kidney International. 2026; 109: 1091-1094.
IgA肾病既往发病机制研究长期以半乳糖缺陷型IgA1(Gd-IgA1)为核心,形成经典“四重打击”学说,IgA2被视作无病理作用的被动共沉积分子。但越来越多临床观察、动物实验与临床试验暴露出传统模型的固有缺陷,仅靠Gd-IgA1无法完整解释IgA肾病发病与进展。近期系列研究证实,IgA2广泛沉积于IgA肾病患者肾小球系膜区,通过激活补体、招募炎性细胞、加重肾小管间质纤维化放大肾脏损伤,完善了IgA肾病发病机制框架。本文梳理传统学说的矛盾,对比IgA1与IgA2结构及免疫功能差异,阐述IgA2致病证据,介绍整合两种IgA亚型的新型发病模型,并探讨该理论对精准治疗的指导价值。
一、传统IgA肾病“四重打击”模型的局限性
经典“四重打击”理论将Gd-IgA1作为IgA肾病核心致病分子:IgA1铰链区糖基化异常,生成具备自身抗原属性的Gd-IgA1;机体随之产生抗聚糖抗体,二者结合形成循环免疫复合物,沉积于肾小球系膜区,最终诱发肾小球炎症损伤。该理论能够解释患者肾小球IgA1沉积、血清Gd-IgA1升高两大特征,长期指导领域内基础与临床研究。
但近年多项证据证明,单纯Gd-IgA1不足以诱发IgA肾病。其一,血清Gd-IgA1升高无疾病特异性,健康人群、患者无症状亲属均可出现该指标上升;体外实验中单体Gd-IgA1无法诱导系膜细胞损伤。其二,基因编辑人源化小鼠模型中,B细胞特异性高表达Gd-IgA1,却未出现肾小球IgA沉积与肾脏病变。其三,felzartamabⅡ期临床试验显示,药物可长期改善蛋白尿,但停药后血清Gd-IgA1迅速恢复基线,肾脏保护效应持续存在,说明免疫复合物沉积才是肾脏损伤关键,而非Gd-IgA1本身。
供肾病理研究进一步佐证模型缺陷:5%~15%西方人群、40%东亚供肾存在无症状系膜IgA沉积,且沉积物仅含IgA1,无任何肾脏病理损伤;同时部分动物实验可脱离Gd-IgA1通路诱导类IgA肾病病变。上述证据表明,IgA肾病发病存在IgA1以外的关键调控因子,IgA2的病理作用亟待重新评估。
二、IgA1与IgA2结构、糖基化及免疫功能差异
人体IgA分为IgA1、IgA2两种亚型,二者铰链区结构、黏膜分布、糖基化修饰与促炎能力存在本质区别,直接决定其在肾病中截然不同的病理角色。
分布层面,血清IgA以IgA1为主(80%~90%);IgA2集中分布于菌群密集、蛋白酶丰富的肠道黏膜;上呼吸道、上消化道黏膜IgA1占优(65%~70%),结肠黏膜IgA2占比达65%,是IgA2主要合成位点。黏膜B细胞产生多聚IgA,经上皮转运形成分泌型IgA,分泌型IgA2耐受强酸与蛋白酶降解,稳定调控肠道菌群、中和致病菌毒素,构成肠道黏膜防御屏障。
结构差异集中于铰链区:IgA1铰链区长且柔性,富含O-糖基化位点,抗原识别范围广,但易被细菌蛋白酶水解;IgA2铰链区短,缺失O-糖基化位点,分子刚性强,抗蛋白酶切割,适配肠道恶劣环境。糖基化修饰调控炎症活性:IgA1唾液酸化水平高,循环单体IgA1整体呈抗炎效应;IgA2唾液酸化程度低,聚集状态下可强烈刺激巨噬细胞、中性粒细胞释放促炎因子。人为去除IgA1唾液酸后,其促炎活性接近IgA2,证明糖基化是亚型免疫病理活性的核心调控因素。
补体激活能力是二者最关键的病理分界点。补体旁路、凝集素通路激活是IgA肾病肾损伤核心驱动力,且两种亚型激活效率差异显著。单体IgA1通过CD89受体传递抑制性炎症信号;IgA2促炎效应不依赖经典CD89通路,多聚体IgA2可高效激活补体旁路与凝集素通路,诱导中性粒细胞胞外诱捕网与大量炎症因子释放。过敏性紫癜肾炎活检可见IgA1、IgA2共沉积,IgA2与凝集素通路激活、不良预后直接相关,侧面证实其促损伤作用。
三、IgA2具备独立致病放大效应的循证证据
早期研究受抗体交叉反应等技术限制,难以稳定检出肾小球IgA2,长期将其归为无意义共沉积产物。2026年Li等人发表的多中心临床与转基因小鼠研究,提供了IgA2主动介导肾损伤的直接因果证据。
临床研究纳入161例中国多中心IgA肾病肾活检标本,免疫荧光显示98%以上患者系膜区存在IgA2沉积,包含IgA2(m1)、IgA2(m2)两种同种异型,前者多见于白种人,后者为亚洲、非洲人群优势亚型。质谱定量显示肾小球总沉积IgA仍以IgA1为主,IgA2含量仅为IgA1的1/50,但具备明确病理价值;IgA肾病患者血浆IgA2较健康对照升高约30%。肾小球IgA2沉积荧光强度与病理损伤程度显著正相关:IgA2高沉积患者系膜增生、肾小管萎缩间质纤维化更严重,肾脏C3激活水平升高,估算肾小球滤过率更低,证明IgA2沉积直接推动肾脏慢性病变。
为建立因果关系,研究构建人源化IgA1、IgA2特异性转基因小鼠,经干酪乳杆菌细胞壁提取物刺激模拟黏膜免疫紊乱。结果显示,IgA2转基因小鼠补体活化、巨噬细胞浸润、肾小管萎缩程度均显著高于IgA1小鼠,证实IgA2并非被动随IgA1沉积,而是主动推动肾脏炎症与纤维化。该研究存在一定局限:临床队列仅覆盖中国人群,结论需多种族验证;人源化小鼠无法完全复刻人类自发性IgA肾病病程,但仍是目前IgA2致病最核心体内证据。
综上,IgA肾病“四重打击”发病模型需进一步更新完善。Gd-IgA1依旧是疾病核心,但现有证据证明IgA2同样发挥关键作用。肠道来源IgA2可进入血液循环,形成免疫复合物,与含IgA1的复合物共同沉积于肾小球系膜区。IgA2沉积机制、免疫复合物组分仍需借助新技术深入解析。图1展示了含IgA2循环免疫复合物的潜在作用模式:IgA2沉积后,可强效激活补体、招募巨噬细胞,加速肾小球与肾小管间质损伤。后续仍需探索IgA2免疫复合物的形成过程、抗原组分;IgA2可能通过黏膜移位大量入血,或由骨髓、淋巴组织异常过量合成。因此IgA肾病不只是IgA1糖基化异常、抗Gd-IgA1抗体生成类疾病,黏膜IgA亚型失衡同样会驱动肾脏进行性损伤。
图1.IgA1和IgA2对IgA肾病贡献的整合模型
四、IgA2致病理论对临床治疗的指导价值
现有IgA肾病治疗多广谱抑制B细胞或阻断下游炎症,IgA2致病机制的明确,为分层精准治疗提供四类新思路。
第一,调控黏膜免疫与肠道菌群的疗法,可优先减少IgA2合成;
第二,靶向补体的药物,尤其是作用于凝集素、旁路通路的制剂,可特异性缓解IgA2介导的肾损伤;
第三,开发Fcα受体靶向药物,可调控IgA1、IgA2与髓系细胞的相互作用;
第四,检测IgA亚型可优化风险分层,精准识别IgA2炎症主导、进展风险更高的患者。
该理论并未否定“四重打击”学说,而是补充完善原有发病框架:IgA1启动免疫复合物生成,IgA2则调控IgA肾病炎症与纤维化进展。重新认识IgA2不会削弱Gd-IgA1的核心地位,反而将黏膜免疫、补体激活、亚型特异性炎症整合为一套更完整的发病机制。随着治疗手段不断发展,充分理解IgA1、IgA2的双重作用,是实现从单纯抑制症状到真正逆转病程、精准靶向致病IgA亚型的关键。
五、总结与展望
长久以来IgA肾病研究局限于IgA1单一亚型范式,IgA2独特的分子结构、黏膜分布与强促炎促纤维化能力,使其从“无关旁观者”转变为重要致病放大器。多中心活检证实绝大多数IgA肾病存在肾小球IgA2沉积,沉积水平与病理损伤、肾功能预后直接相关;人源化动物模型确立IgA2加重肾损伤的因果关系,搭建起IgA1、IgA2协同致病的整合模型。该理论打通黏膜免疫、IgA亚型生物学、补体激活与肾脏纤维化的关联,开辟多条全新治疗靶点。
参考文献:Seikrit C, Floege J. Kidney International. 2026; 109: 1091-1094.
- 推荐文章
邱福宇教授:启动更早,达标更严?欧美最新高血压指南的策略分歧与临床启示
母胎安全的边界与突围!当慢性肾病遇上妊娠,CKD患者妊娠管理全攻略
Jürgen Floege教授最新述评|超越半乳糖缺陷型IgA1:重新审视IgA2在IgA肾病中的致病驱动作用
诺兰多西呱惊艳亮相,CKD残余风险迎来“终结者”?——ERA前主席解读心肾疾病管理方向
路万虹教授:基于2026 KDIGO指南更新的肾性贫血诊疗进展
虚弱不仅是结局,更可能推动糖尿病肾病进展?
研究者解读:SGLT2抑制剂治疗Alport综合征的新证据
ERA 2026前沿 | 突破诊疗壁垒,迈向全程管理:构建以SGLT2i为核心的CKD早筛早诊早治新格局
崔兆强教授:利尿剂——高血压治疗中难以替代的“基石”与“放大器”
FDA就暂不批准碳酸氧镧治疗透析慢性肾脏病患者高磷血症的适应证发出完整回应函
CKD管理新证据:司美格鲁肽改善生活质量,阿司匹林用于心血管疾病一级预防不推荐
CKM视角下的心血管一级预防:为何司美格鲁肽被推至前沿?
靶向补体治疗免疫性肾病的临床研究进展
佟倩教授:高血压治疗,指南与临床的鸿沟如何跨越?
无尿背后的致命陷阱!肾皮质坏死,比急性肾损伤更凶险的沉默杀手
对话何志凌教授:从“降压达标”到“靶器官共护”,国产原研ARNI如何重塑高血压全生命周期管理?
肾实质性高血压诊疗路径:从机制解析到精准靶目标管理,再到药物选择的肾保护策略
高血压精准管理新动向:关注难治性高血压患者中皮质醇增多症的筛查丨ACC.26
家族里多人早发痛风?小心一种常染色体遗传病:尿调节素相关肾病会悄悄毁肾
推翻激素假说:炎症是慢性肾脏病女性生育力受损的“隐藏推手”——一项观察性蛋白质组学与遗传流行病学研究探索
双靶点协同护肾心——醛固酮合酶抑制剂联合SGLT2 抑制剂的BaxDuo系列临床研究项目稳步推进
久坐越少越好?最新研究带来新发现:每天约4小时久坐风险最低
葡萄牙学者发布初发肾病综合征患者5年肾衰竭风险预测模型
早干预、规范治、新药破局——慢性肾脏病心肾保护新策略解读
C4d沉积预警IgA肾病患者肾衰竭,凝集素通路激活提示预后不良
第二届肾小球疾病中外大咖面对面(CIG):深耕肠道黏膜靶向治疗,筑牢IgA肾病对因治疗、保护肾功能基石地位
OCN 2026丨臧秀娟教授:非透析患者HK的诊疗新进展——从隐匿诊断到精准治疗的“破局”
张宏/吕继成教授深度专访:首个中国原研SGLT2i恒格列净联合内皮素受体拮抗剂证据荣登JASN,破解临床联合用药困局
肾例明鉴丨68岁老人反复鼻塞咳血+肾功能急跌!警惕这种“伪装”成肿瘤的自身免疫病
学术纵横|“满堂亮”是IgA肾病肾小球坏死的可靠标志物,不同补体抑制剂感染风险存异
李冰教授:IgA肾病机制及治疗新进展——未来已来
【会议预热】大咖云集,共探前沿|第二届肾小球疾病中外大咖面对面(CIG)学术研讨即将启幕!
CCBPC 2026 | 梁伟教授:用药越多,疗效越差?CKD药物相互作用的临床警示
中国2026 CKM综合征评估、诊断、治疗专家共识重磅发布,CKM 0~4期管理路径尽收眼底
边波教授:从血压控制到心血管风险防控,步入高血压综合管理新时代
盘点6月肾脏领域新进展:多款新药获批+多项重磅研究数据收入囊中,学习休闲两不误
2026年度SCI影响因子正式发布,肾脏病与泌尿领域最新TOP 10期刊排名揭晓,哪些期刊成功上榜?
余静教授:高血压管理新风向——从疾病负担到精准治疗的全面革新
CCBPC丨阳晓教授:进展至无尿,腹透还值得坚持吗?
7月4日-5日 | 广东省医院协会肾脏病防治与血液净化中心管理专业委员会换届选举会议暨2026年学术年会
临床必备!2026版中国CKD筛查、诊断及治疗指南32条推荐意见、12张表格、1张流程图速览
NDT:三大国际权威指南更新对比,狼疮性肾炎迎来靶向新时代
蔡军教授:心血管病诊疗向治愈进阶、向预防前移、向舒适升级
CCBPC 2026|彭晖教授:腹膜透析影响心血管系统的多维度解析
肾科领域迎来新突破!DMX-200推进至全球Ⅲ期临床研究,FSGS治疗有望打破僵局
中国创新药的破局之作:揭秘全球第二款ARNI类药物沙库巴曲阿利沙坦的研发征程
学术纵横|血清PLA2R抗体检测存在局限性,奥妥珠单抗治疗难治性pMN疗效确切
《补体相关性肾病诊断和治疗专家共识》要点概览
UACR降幅超40%!中国迄今最大规模非奈利酮真实世界研究在ADA2026首次公布
韩飞教授:联合免疫靶向治疗急进性肾炎的临床探索与研究进展
沪上聚贤论肾道,东方盛会谱新篇——第十六届东方肾脏病学会议开幕
肾例明鉴丨隐匿在民间偏方里的肾衰陷阱!“朱砂莲”暗藏马兜铃酸剧毒,男子从服药到肾衰仅十余天
超万人注册参会、新药爆发、异种移植可期——ERA秘书长详解第63届ERA大会重磅进展
时隔4年,2026年ADA/EASD 2型糖尿病高血糖管理共识声明草案公布 | ADA2026
ERA专家访谈 | 范秋灵教授:补体抑制剂助力蛋白尿深度缓解,口服盐酸兰诺可泮治疗lgA肾病12周达标率高达40%