
- 首页 > 正文
肾域华章 | 北大医院张宏教授团队探索FHR-1在IgA肾病肾损伤中的双重作用
发表时间:2026-02-02 11:40:20
编者按
IgA肾病(IgAN)是全球最常见的原发性肾小球肾炎,其发病机制复杂,补体系统在其中扮演关键角色。近年来,补体调节蛋白家族成员之一——因子H相关蛋白1(factor H-related protein 1,FHR-1)被认为与IgAN的发生、发展密切相关。然而,FHR-1的具体致病机制尚未阐明。北京大学第一医院张宏教授团队在Kidney International Reports发表的最新研究,揭示了FHR-1在IgA肾病肾损伤中的“激活-炎症”双重作用,并提出其截短片段FHR-1SCR1-2具有潜在的治疗前景。

一、IgA肾病的补体异常与FHR-1研究背景
IgA肾病的特征为系膜区IgA1免疫复合物(IgA1-IC)沉积,常伴补体C3共沉积,引起系膜细胞增生及炎症细胞浸润。据流行病学数据显示,约40%的患者在确诊后20~30年内进展为终末期肾病(ESKD)。临床与实验研究均证实,补体活化程度与肾脏病理损伤严重程度及预后密切相关。
补体可通过三条途径被激活:经典途径、凝集素途径和替代途径,其中替代途径在IgAN中占主导地位。多达87%的患者可见系膜Bb沉积,提示替代途径持续激活。替代途径抑制剂伊普可泮(Iptacopan)的Ⅱ期随机对照研究证实,抑制替代途径可显著降低尿蛋白水平,从而验证了这一机制的临床重要性。
全基因组关联研究(GWAS)在1q32位点发现补体因子H相关蛋白1(CFHR-1)和补体因子H相关蛋白3(CFHR-3)的双基因缺失与IgAN的保护作用相关,而循环FHR-1升高及FHR-1/FH比值增加与疾病进展显著相关。这些发现提示:FHR-1可能通过调控补体活化及炎症反应参与IgAN的发病过程。
二、FHR-1的双重生物学功能:补体失调与炎症触发
FHR-1缺乏N端SCR1–4调节结构域,使其无法像因子H(FH)一样发挥抑制作用,反而可竞争性结合配体,促进补体持续激活。此外,FHR-1还具有“非经典”炎症效应:可结合坏死细胞膜上的氧化脂质(如丙二醛MDA表位),并与单核细胞表面受体EMR2相互作用,激活NLRP3炎症小体,从而诱导IL-1β和IL-18释放。FHR-1在ANCA相关血管炎和动脉粥样硬化等炎性疾病中同样升高,提示其炎症介导特性具有广泛生理病理意义。
三、研究设计与实验模型
研究团队通过体外及体内模型系统性探索FHR-1在IgAN肾损伤中的作用机制:
体外实验:使用患者来源的cIgA1-IC刺激人肾系膜细胞,模拟IgA沉积;并观察重组FHR-1与受损系膜细胞的结合、补体活化及与单核细胞共培养后的炎症反应。
体内实验:采用肝脏特异性表达人FH与FHR-5的转基因小鼠(hFH-FHR5),通过腹腔注射Lactobacillus casei细胞壁提取物(LCWE)诱导IgA沉积。随后给予FHR-1或其片段FHR-1SCR1-2处理,以评估肾脏炎症与补体沉积变化。
四、主要研究结果
1. FHR-1与受损系膜细胞结合能力增强
cIgA1-IC刺激后,FHR-1对系膜细胞的结合显著增加。进一步实验表明,这种结合并非通过C3b介导,而是依赖细胞表面MDA表位;使用抗MDA抗体可明显抑制FHR-1与肾小球系膜细胞的结合,提示MDA可能是肾小球系膜细胞上FHR-1的结合位点(图1,D)。值得注意的是,FHR-1的SCR1-2结构域可独立结合系膜细胞,并轻度抑制全长FHR-1肾小球系膜细胞的结合(图1,E-F),提示该区域是关键识别结构。上述结果表明,在cIgA1-IC损伤的肾小球系膜细胞上,FHR-1可通过SCR1-2结构域与MDA发生相互作用。
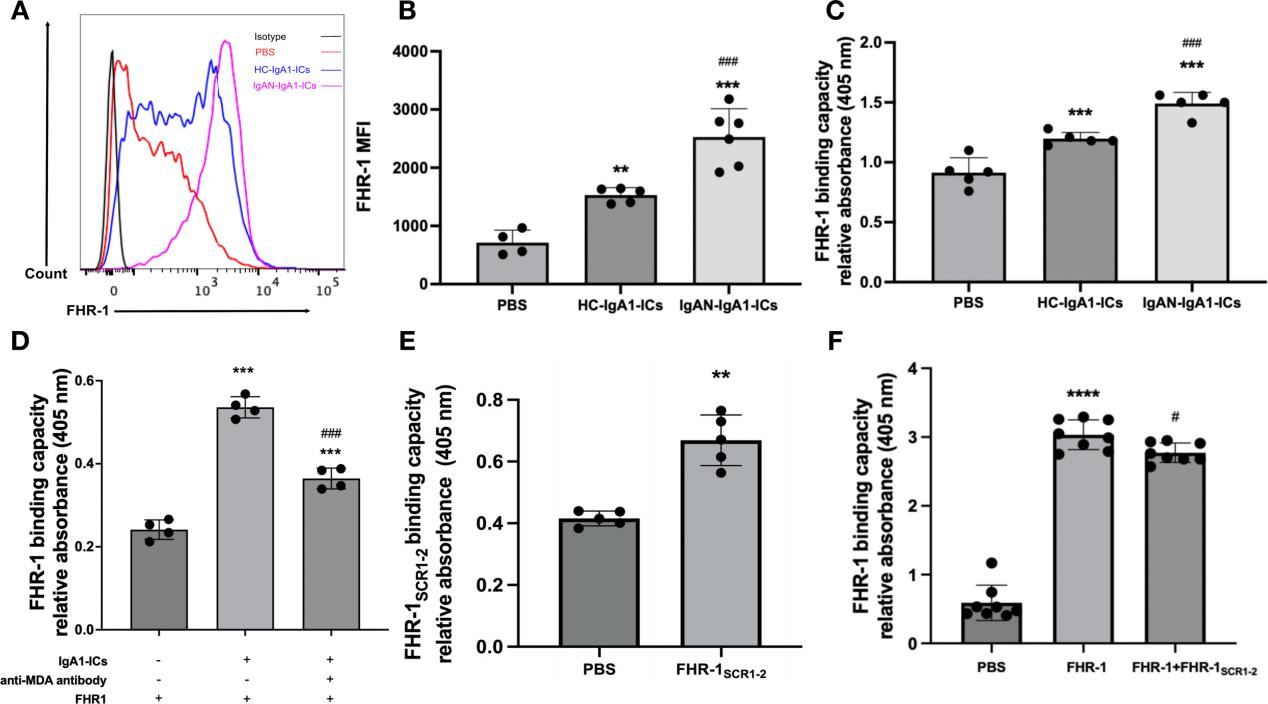
图1. cIgA1-IC刺激后FHR-1通过SCR1-2结构域与肾小球系膜细胞结合
2.系膜细胞结合的FHR-1激活单核细胞炎症反应
在系膜-单核细胞共培养体系中,FHR-1可显著诱导IL-1β和IL-18分泌,单独培养系膜细胞时不出现此效应,提示主要炎症来源于单核细胞。进一步研究发现,抑制NFκB(核因子κB)、NLRP3(NLRP3炎性小体)和caspase-1(半胱天冬酶-1)可几乎完全阻断IL-1β和IL-18的分泌(图2,C-D),这一结果进一步支持了上述结论。
此外,研究还观察到,在FHR家族成员中,仅FHR-1在共培养体系中表现出炎症诱导效应,而FHR-2、FHR-3、FHR-4、FHR-5及FH均无类似效应。
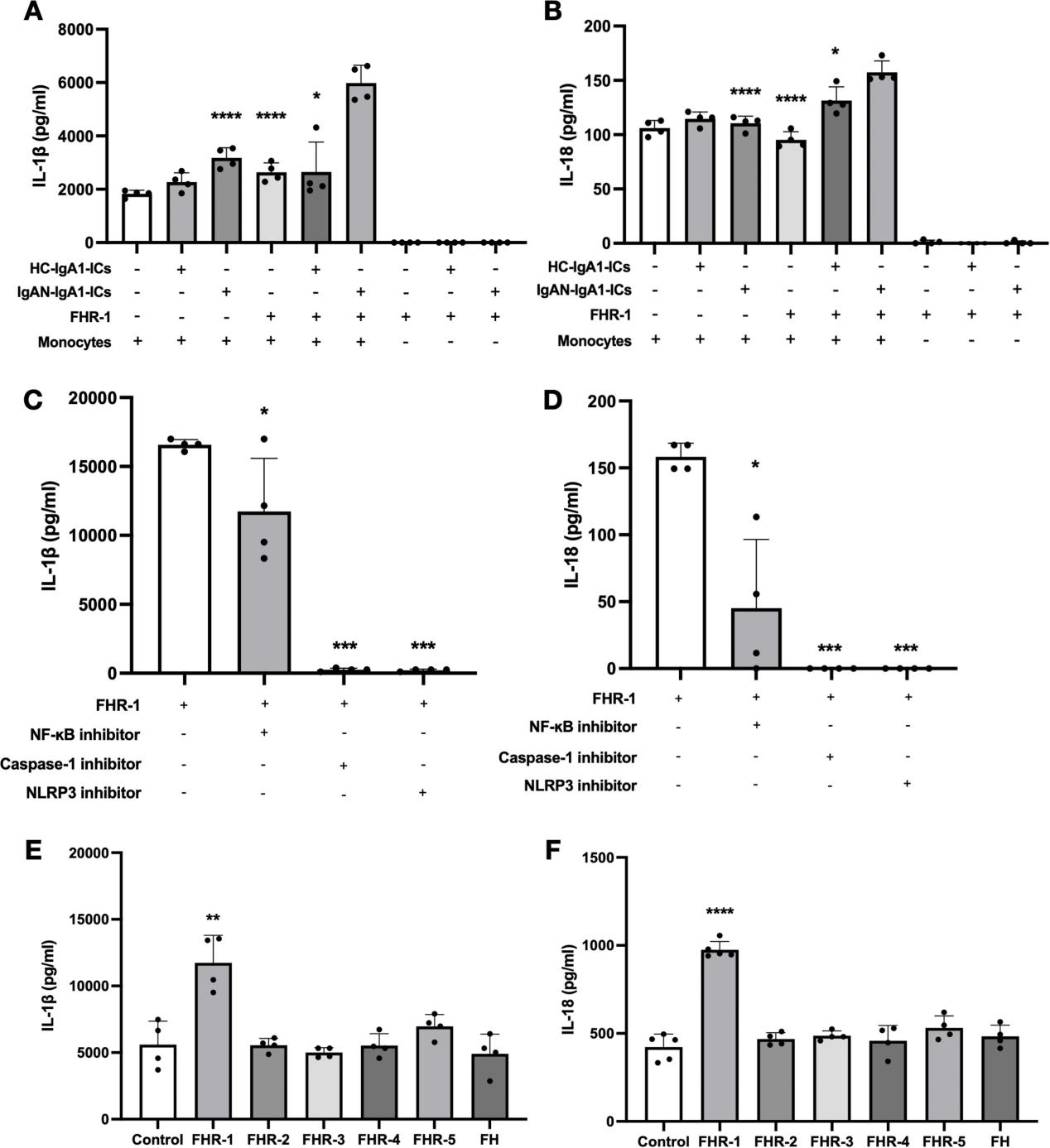
图2. 结合于肾小球系膜细胞的FHR-1诱导单核细胞炎症
3.FHR-1通过双途径诱导炎症:直接结合与补体依赖机制
FHR-1直接触发单核细胞炎症:FHR-1与单核细胞受体EMR2结合可直接触发炎症反应。使用FHR-1SCR1-2干扰FHR-1与肾小球系膜细胞的结合(图3,A-B),或者使用EMR2抗体FHR-1与单核细胞上EMR2受体的结合(图3,C-D),均显著降低IL-1β和IL-18水平。
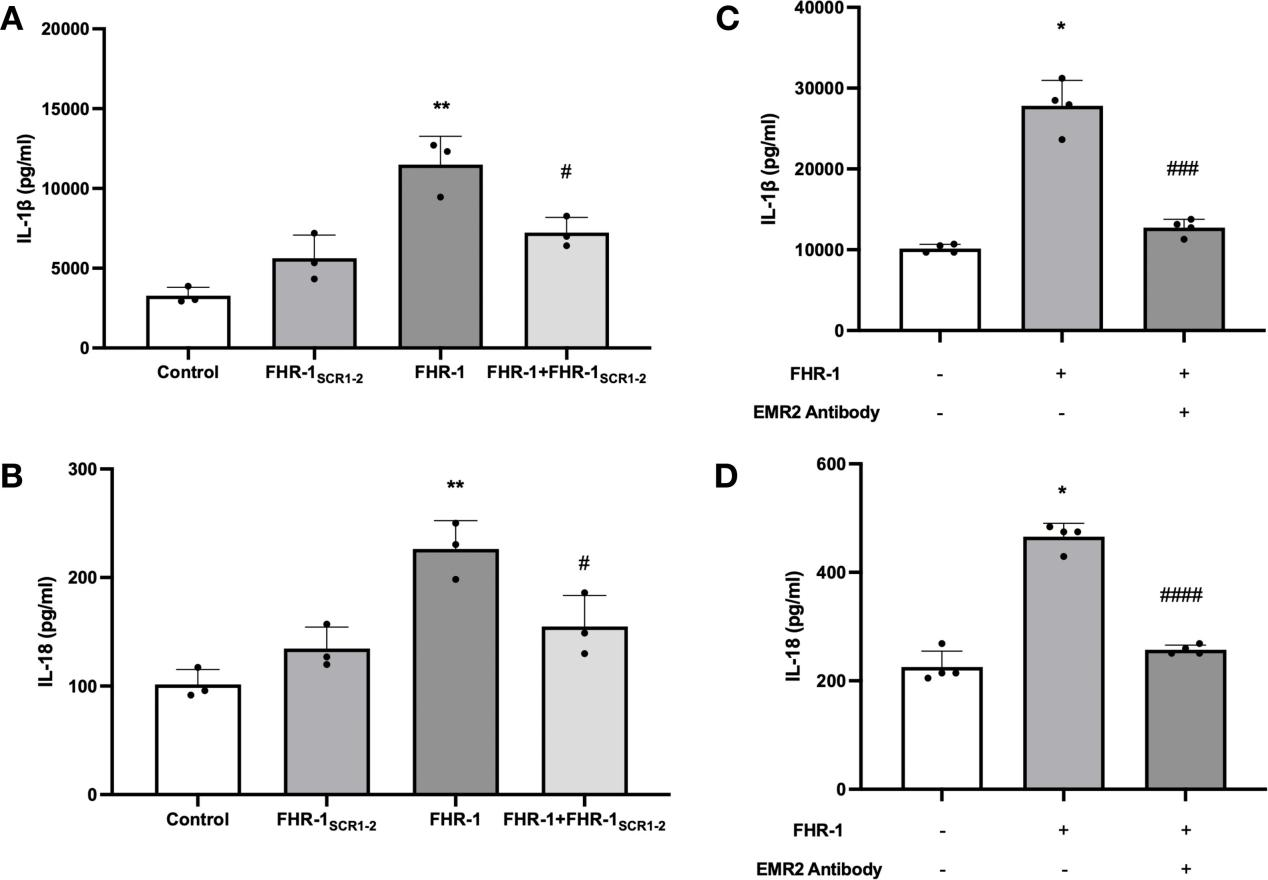
图3. 抑制FHR-1与单核细胞或肾小球系膜细胞的相互作用可减轻单核细胞炎症
FHR-1通过补体激活间接触发单核细胞炎症:FHR-1可增强IgA-IC诱导的补体C3c沉积(图 4,B-C)。使用去除不同补体成分的血清后,炎症反应显著下降,其中去除替代途径关键因子FB或FP的抑制最明显,提示替代途径在FHR-1介导的炎症中占主导。进一步应用C3aR及C5aR拮抗剂后,炎症反应同样减弱,说明C3a-C3aR与C5a-C5aR轴在此过程中发挥关键作用(图 5,A-D)。
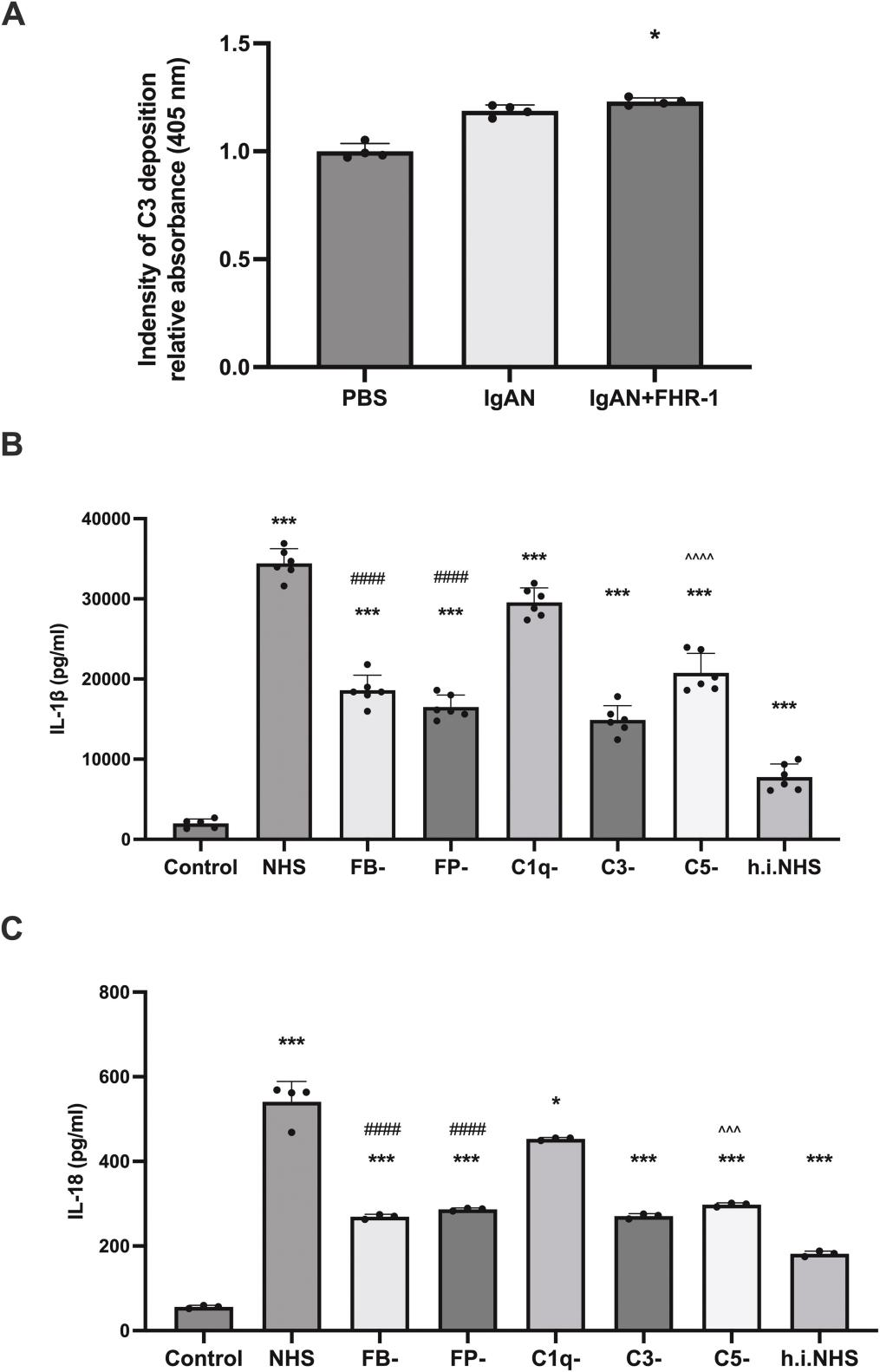
图4. FHR-1通过补体激活间接触发单核细胞炎症
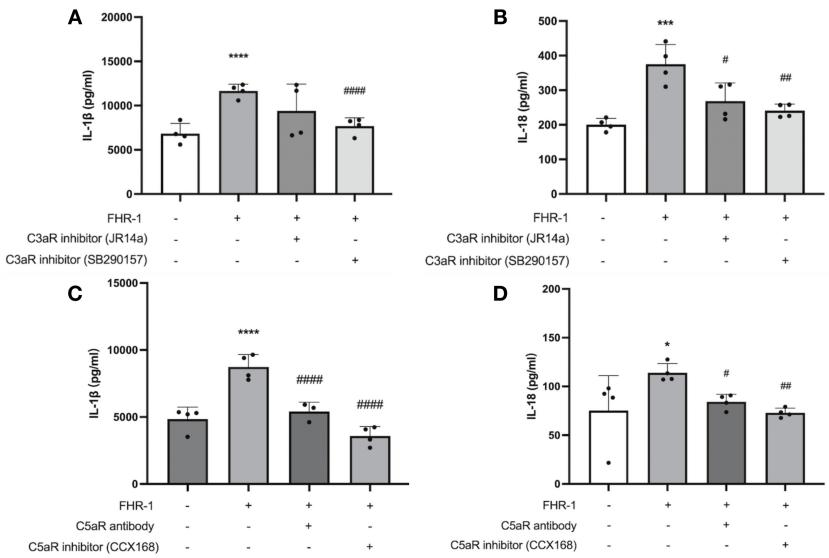
图5. 在系膜细胞-单核细胞共培养体系中,抑制C3aR和C5aR可减轻FHR-1诱导的单核细胞炎症
4.体内验证:FHR-1加重IgA沉积小鼠的补体激活与炎症
在IgA沉积模型小鼠中,腹腔注射FHR-1后,肾小球C3沉积面积与强度显著增加,同时F4/80+巨噬细胞及Ly6G+中性粒细胞大量浸润。与之相比,共注射FHR-1SCR1-2可明显缓解补体沉积及炎症细胞浸润(图6),表明该截短片段具有拮抗FHR-1致病效应的潜力。
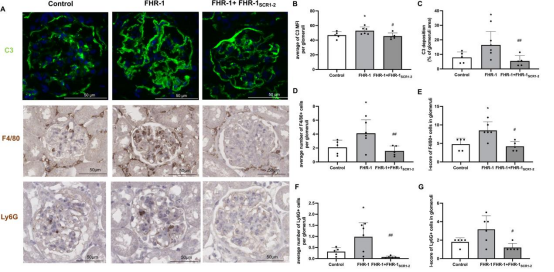
图6. FHR-1 增强肾小球IgA肾病沉积小鼠的肾小球补体激活与炎症反应
五、机制解析与临床启示
研究揭示,FHR-1在IgAN中具有“双重角色”:
补体调节失衡作用(间接):通过与FH竞争C3b结合位点,促进补体持续激活及炎症介导因子的释放;
炎症直接诱导作用(直接):通过SCR1-2区与受损系膜细胞结合,并与单核细胞EMR2受体互作,激活NLRP3炎症小体。
两种机制共同加重局部炎症反应和肾小球损伤。更重要的是,FHR-1SCR1-2能与FHR-1竞争结合位点,从而阻断其直接促炎作用并削弱补体激活,表现出一定的肾脏保护效应,为未来药物开发提供新方向。
研究亦提示,在IgAN的精准治疗策略中,单纯抑制下游C5可能不足,靶向C3及其上游调控蛋白(如FHR-1)或可获得更全面的抗炎获益。
六、研究局限与展望
作者指出,本研究使用的LCWE模型虽能模拟IgA沉积,但小鼠IgA结构与人类不同,缺乏O-糖基化异常,因此仍难完全再现人类IgAN的病理机制。此外,FHR-1SCR1-2在体内抑制补体的具体分子机制尚待深入研究。未来有必要在更符合人类IgAN特征的模型中验证FHR-1干预的治疗效果,并进一步探索其与其他补体抑制剂的联合策略。
七、结论
综上所述,FHR-1在IgA肾病中通过两种途径加重肾脏炎症与损伤:一方面,它促进补体系统的过度激活,从而间接诱导炎症;另一方面,IgA沉积导致的系膜细胞损伤为FHR-1结合提供了新靶位,进一步触发单核细胞-炎症小体反应。FHR-1截短片段FHR-1SCR1-2可有效阻断这一过程,提示其有望成为IgAN新型补体-炎症双通路干预的候选治疗分子。
来源:Kang Y, Li X, Yuan X, Shi S, Lv J, Zhu L, Zhang H, Dual role of FHR-1 in IgA nephropathy kidney injury, Kidney International Reports (2025), doi: https://doi.org/10.1016/j.ekir.2025.09.047
- 推荐文章